
Biopharmaceutical Quality Control: A Comprehensive Guide To QA And QC Processes
Quality assurance (QA) and quality control (QC) are crucial to biopharmaceutical products' safety, efficacy, and regulatory compliance. Integrating QA/QC into a comprehensive quality management system (QMS) allows biomanufacturers to mitigate risks, improve traceability, and consistently produce high-quality therapeutics.
Table Of Contents:
- What Is The Importance Of QA And QC In Biopharmaceutical Manufacturing?
- What Are The Key Differences Between QA And QC?
- What Regulatory Frameworks Govern QA/QC?
- How Are QA/QC Processes Integrated Throughout The Biopharmaceutical Lifecycle?
- What Analytical Methods Are Used In Biopharmaceutical QC?
- What Are the Best Practices For Quality Assurance Systems?
- What Common Challenges Do Biopharmaceutical Companies Face When Implementing A QMS?
- What Emerging Trends Are Revolutionizing Biopharmaceutical QA/QC?
- Conclusion
- Frequently Asked Questions (FAQs)
What Is The Importance Of QA And QC In Biopharmaceutical Manufacturing?
QA is a proactive, systematic framework that minimizes variability and prevents defects throughout the entire drug manufacturing lifecycle. QC, in contrast, is a reactive process that inspects, tests, and verifies that biological drug products meet predetermined quality standards.
QA and QC are of the utmost importance in biopharmaceutical manufacturing. Biological systems are inherently complex and variable, creating significant safety risks if defective products are given to patients. Also, strict regulatory requirements from agencies like the FDA and EMA demand a robust QA/QC strategy.
What Is Quality Assurance?
QA sets quality standards, policies, and procedures for designing and implementing comprehensive QMSs. In short, a QA system includes audits and reviews, provides employee training, and manages documentation and change control.
What Is Quality Control?
QC complements QA by ensuring the biopharmaceutical product is safe and effective. QC processes include batch inspections, product sampling, laboratory tests of raw materials, in-process samples, and final product checks. This system also validates manufacturing processes and serves as the final step so that only the highest quality products reach patients.
This comprehensive guide explains the fundamentals of QA and QC and their role in bioprocessing and covers testing procedures, regulatory requirements, best practices, and emerging industry trends.
What Are The Key Differences Between QA And QC
QA and QC are commonly grouped in biopharmaceutical manufacturing strategies, but they are distinct, complementary systems that protect drug quality, safety, and efficacy. QA establishes preventive measures and processes, while QC verifies that these measures are effective by testing and inspecting the final product. Thus, they differ in their approach, timing, and focus.
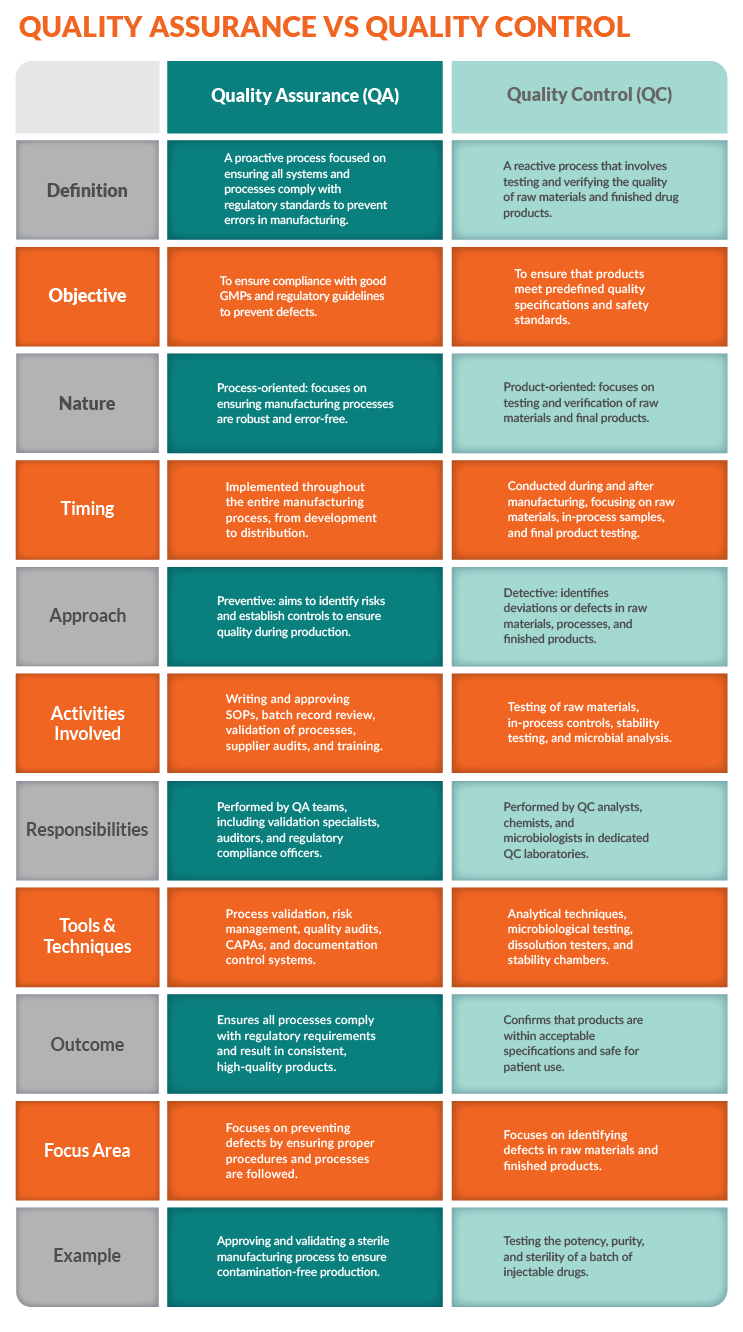
The Difference Between Process And Product Orientation
QA is process-oriented, establishing and maintaining systems and procedures to prevent defects and ensure quality throughout the entire production lifecycle via:
- Defining quality standards and specifications
- Implementing QMSs
- Running audits and reviews
- Providing employee training
- Managing documentation and change control
QC, on the other hand, is product-oriented, inspecting and testing the final product or its components to detect and correct issues by:
- Performing batch inspections and product sampling
- Conducting laboratory testing on raw materials, in-process samples, and final products
- Validating manufacturing processes
Taking A Proactive Vs. A Reactive Approach
QA proactively prevents defects by ensuring adherence to established processes and standards. It builds quality into the product from the start by creating a framework that minimizes variability.
QC, on the other hand, is reactive, detecting and correcting defects after production. It is the final "gate" to ensure only safe, high-quality products reach patients.
How Timing And Scope Differ Between QA And QC
QA activities encompass the entire production process, from planning to execution. They are ongoing throughout the product's lifecycle.
However, QC activities typically occur at the end of the production process when the final product is ready for inspection and testing and are more specific to individual products or batches.
How Do QA And QC Treat The System Vs. Parts?
QA procedures are holistically applied across the system, establishing and maintaining processes to ensure overall quality.
Meanwhile, QC concentrates on individual system parts, inspecting and testing specific products or components to detect issues and ensure they meet established quality specifications.
How QA And QC Work Together
QA and QC teams collaborate closely to maintain product integrity and regulatory compliance in biopharmaceutical manufacturing. QA establishes quality systems, procedures, and standards, while QC implements testing and inspection protocols. QA conducts audits and reviews QC data to ensure adherence to quality standards. QC provides feedback on process performance and product quality, enabling QA to refine procedures. Together, they investigate deviations, implement corrective actions, and continuously improve processes.
What Regulatory Frameworks Govern QA/QC?
Worldwide, regulatory bodies carefully oversee the manufacture of biological drug products. These guidelines shape QA/QC practices to protect biopharmaceutical products' consistency, safety, and efficacy.
Overview Of International Regulations And Guidelines
Regulatory agencies such as the FDA, EMA, ICH, and WHO provide a framework for bioprocessing manufacturers to establish comprehensive QA/QC systems. These guidelines have several elements in common, such as:
- Risk-based approach to quality management
- Emphasis on process understanding and control
- Continuous improvement and lifecycle management
- Robust documentation and traceability
- Validation of processes, equipment, and analytical methods
FDA
The FDA emphasizes risk-based approaches and continuous improvement in quality systems in the following documents:
- 21 CFR Parts 210 and 211: Good Manufacturing Practice regulations for drugs and biologics
- 21 CFR Part 600: Biological Products Regulations
EMA
The EMA has similar guidelines, including these key regulations:
- EudraLex Volume 4: EU Guidelines for Good Manufacturing Practice for Medicinal Products for Human and Veterinary Use
- Specific guidelines for biological medicinal products
ICH
ICH’s harmonized guidelines help ensure global consistency and are widely adopted by regulatory agencies worldwide:
- ICH Q10: Pharmaceutical Quality System, which provides a model for an effective QMS
- ICH Q8: Pharmaceutical Development
- ICH Q9: Quality Risk Management
WHO
The WHO also provides global standards and recommendations, such as:
- WHO Good Manufacturing Practices for Biological Products
- Guidelines on quality, safety, and efficacy of biotherapeutic products
Understanding Good Manufacturing Practices And Good Laboratory Practices
GMPs and GLPs are required by international regulatory bodies which emphasize risk assessment and management. These agencies strive for continuous improvement, adapting to new technologies and modalities as the industry evolves. Regulatory bodies combine guidelines, such as those listed above, with education and enforcement tactics, including:
Training And Education
Typically, regulatory bodies provide training resources and workshops to help industry professionals understand and implement GMP and GLP standards.
Certification Programs
Some agencies offer voluntary certification programs for GMP compliance, such as the EMA's Certification of Suitability to the Monographs of the European Pharmacopoeia.
Inspections
Manufacturing facilities and laboratories undergo regular inspections that assess GMP and GLP compliance.
Enforcement Actions
Failure to comply with regulations can result in warning letters, product recalls, or even suspension of manufacturing licenses.
What GMPs And GLP Are Unique To Biopharmaceutical Manufacturing?
While GMPs and GLPs apply broadly across pharmaceutical manufacturing, biopharmaceutical manufacturing carries unique aspects that require specific guidelines, such as:
Aseptic Processing
Biopharmaceuticals are highly susceptible to contamination, necessitating the use of aseptic processing. Regulations such as the FDA's "Guidance for Industry: Sterile Drug Products Produced by Aseptic Processing" provide precise requirements for sterile manufacturing.
Cell Culture And Fermentation
Cell culture and fermentation processes unique to bioprocessing are carefully controlled, including cell line development, media preparation, and bioreactor operations.
Viral Safety
Specific guidelines protect biopharmaceutical safety, including viral clearance studies and validation of viral removal/inactivation steps.
Process Analytical Technology (PAT)
In particular, the FDA encourages PAT in biopharmaceutical manufacturing to ensure consistent product quality.
QbD
QbD approaches are especially relevant to biopharmaceutical manufacturing due to the inherent complexity of biological processes.
Biosafety Levels
Biopharmaceutical labs often require higher biosafety levels, with specific guidelines for handling potentially hazardous materials.
Environmental Monitoring
Stringent environmental monitoring is typically required for biopharmaceutical manufacturing facilities.
Raw Material Testing
Guidelines for testing and qualifying complex biological raw materials are specific to biopharmaceuticals.
Analytical Method Validation
Biopharmaceuticals commonly require complex analytical methods with specific guidelines for their validation.
Stability Testing
Stability testing guidelines are often more complex for biopharmaceuticals due to the nature of biological products.
Comparability Studies
These studies provide valuable insights when changes are made to biopharmaceutical manufacturing processes.
Single-Use Systems
Implementing and validating single-use systems, popular solutions for biopharmaceutical manufacturers, are strictly regulated.
Key Requirements For Quality Systems, Documentation, And Data Integrity
Quality systems, documentation, and data integrity ensure biopharmaceutical manufacturers maintain high product quality standards, protect patient safety, and meet regulatory expectations.
A robust QMS framework contains clear quality policy and objectives, a comprehensive quality manual, well-defined organizational structure and responsibilities, internal processes and procedures, patient satisfaction monitoring, and continuous improvement mechanisms.
Documentation is vital to maintaining quality standards and traceability. Key documents include SOPs, batch records, quality control records, training logs, equipment maintenance and calibration records, change control documentation, and complaint handling and investigation records. These documents provide a paper trail that adheres to established procedures and facilitates regulatory inspections.
Data integrity is paramount and must conform to the ALCOA principles: Attributable, Legible, Contemporaneous, Original, and Accurate for electronic data management, audit trails, access controls, user management, data backup and recovery procedures, and computerized systems’ validation.
Quality control is confirmed through raw material testing, in-process controls, finished product testing, stability testing, and environmental monitoring. Facilities and equipment must be properly designed, maintained, and validated to ensure consistent product quality, including equipment qualification, calibration, and preventive maintenance programs.
Personnel management entails clearly defined roles and responsibilities, comprehensive training and competency assessment programs, and ongoing education and development opportunities for staff. Risk management strategies, including risk assessment, mitigation procedures, and change management protocols, are necessary to protect product quality and safety.
Compliance with regulatory standards includes adherence to GMP, GLP, and other relevant guidelines. Regular internal and external audits ensure ongoing compliance, and manufacturers must be prepared for impromptu regulatory inspections.
Supplier and materials management includes supplier qualification, monitoring processes, and establishing and adhering to raw material specifications and testing protocols. Regular product quality reviews, including annual assessments and trend analysis of quality data, identify areas for improvement and ensure consistent product quality over time.
Finally, a corrective and preventive action (CAPA) system addresses quality issues promptly and effectively. Procedures are implemented to identify, investigate, and correct quality problems and conduct thorough root-cause analyses to prevent a recurrence. Validation processes, including process validation, cleaning validation, and analytical method validation, safeguard the consistency and reliability of manufacturing processes and quality control measures.
How Are QA/QC Processes Integrated Throughout The Biopharmaceutical Lifecycle?
QA and QC procedures are baked into the entire biopharmaceutical manufacturing lifecycle, from raw material selection to the final product.
How QMS Affects Raw Material Testing And Qualification
High-quality raw materials are necessary to create a potent, safe biopharmaceutical product. Selecting reliable suppliers and rigorously testing their materials up-front ensures regulatory compliance while safeguarding product efficacy and quality.
Supplier Qualification And Management
First, manufacturers must carefully vet any raw material suppliers using the following criteria:
- Initial supplier assessment: The purchasing or procurement department contacts potential suppliers and conducts preliminary evaluations.
- Supplier surveys: Detailed questionnaires, aka paper audits, gather information about potential suppliers’ quality systems and manufacturing processes.
- Sample testing: The manufacturer's quality control lab can request testable samples from prospective suppliers.
- On-site audits: If initial assessments are satisfactory, an on-site supplier audit is scheduled and conducted to verify compliance with quality standards.
- Risk assessment: Risk assessment tools, such as those recommended by ICH Q9, provide guidelines for evaluating multiple suppliers of the same raw materials.
- Ongoing monitoring: Once qualified, suppliers are subject to periodic review and re-qualification to maintain their approved status.
Raw Material Testing Procedures
Next, internal specification sheets are created for each raw material, detailing its required attributes. The raw materials are rigorously and regularly tested to ensure their quality, purity, and suitability, using procedures such as:
- Physicochemical tests: pH, viscosity, particle size, osmolality, etc.
- Identity and purity tests: High-performance liquid chromatography (HPLC), gas chromatography, ultraviolent-visible spectroscopy, Fourier transform infrared, etc.
- Impurity analysis: Heavy metals, residual solvents, organic volatile impurities, etc.
- Microbial testing: Ensures materials are free from contamination.
- Pharmacopeia compliance: Monographs provided by pharmacopeias such as USP, EP, BP, and JP are commonly used to test raw material compliance.
- Critical material focus: Cell culture media components, buffers, and chromatography resins are critical, rigorously tested materials.
- Certificate of analysis (CoA) review: Manufacturers review and verify the CoA provided by suppliers against their testing results.
Integration Into the Overall Quality System
Raw material quality processes are integrated into the QMS to allow for continuous process improvement, risk mitigation throughout the product lifecycle, and enhanced visibility and traceability from design through manufacturing and post-market monitoring. Data integrity and traceability must be preserved throughout the testing process.
QA/ QC’s Roles In Process Development And Validation
QA and QC play vital roles throughout process development and validation and contribute to a robust, holistic quality system.
QA’s Role In Process Design, Development, And Scale-up
QA encourages a proactive approach to integrating quality into the entire manufacturing process and is thus considered from the earliest stages of process design and development. First, QA defines critical process parameters (CPPs) and critical quality attributes (CQAs) based on the target product profile in the process design stage. Risk management principles determine design and control strategies to ensure compliance with cGMP regulations and other relevant guidelines.
As the process is developed, QA operations oversee the design of experiments (DoE) to optimize process parameters, review and approve protocols for characterization studies, and ensure proper documentation of development activities. Next, QA verifies that scale-down models accurately represent the commercial-scale process during scale-up activities. Equipment and facility designs are reviewed to ensure GMP compliance, while technology transfer activities between development and manufacturing are closely monitored.
QC And Process Validation
On the other end of the manufacturing continuum, QC plays a crucial role in process validation, testing and monitoring the manufacturing process to ensure consistency and reliability. QC develops and validates analytical methods to accurately measure CQAs to ensure they are suitable for their intended use throughout the product lifecycle.
QC implements sampling plans and testing procedures for in-process materials and statistical process control tools that monitor process performance and variability during process validation. Next, QC comprehensively tests the finished products, including their identity, purity, potency, and other quality attributes defined in product specifications while stability testing confirms shelf life and storage conditions. In the ongoing verification stage, QC collects and analyzes process data to detect trends and variability and performs periodic reviews to ensure the process remains in control.
Integrating QA And QC
Effective process validation requires close collaboration between QA and QC. QA establishes the overall validation strategy and ensures regulatory compliance, while QC provides the data and analytical support needed to demonstrate process consistency.
How QMS Determines In-Process Controls And Testing
Clearly defining and continuously monitoring CPPs and CQAs enables manufacturers to improve process efficiency, maintain regulatory compliance, and consistently ensure high-quality products throughout production.
The Difference Between CQAs And CPPs
CQAs and CPPs form the foundation of process control and monitoring in biopharmaceutical manufacturing. Both must be monitored in real-time during production to maintain process control and ensure product quality.
Critical Quality Attributes
CQAs are the physical, chemical, biological, or microbiological properties within an appropriate limit, range, or distribution to ensure desired product quality, including purity, potency, stability, and safety attributes.
Critical Process Parameters
CPPs are process parameters whose variability impacts a critical quality attribute and should, therefore, be monitored or controlled, such as temperature, pH, pressure, mixing speed, and flow rates. CPPs are identified through risk assessment, statistical analysis of historical data, and DoE during process development.
Implementing Monitoring Systems
Advanced monitoring systems like the following are necessary to track CQAs and CPPs:
- In-line sensors: Advanced sensors continuously measure critical parameters like pH, temperature, and dissolved oxygen in real time.
- Process analytical technology: PAT tools, such as spectroscopic methods for monitoring product concentration or impurities, provide real-time analysis of critical quality attributes.
- Data management systems: Manufacturing execution systems (MES) or similar platforms collect, store, and analyze real-time process data.
Benefits Of Real-Time Monitoring
Real-time monitoring enables the rapid identification and correction of any process deviations before they impact product quality. These technologies also allow process parameters to be fine-tuned immediately to improve efficiency and consistency while providing valuable data for continuous improvement and process optimization.
Based on real-time monitoring data, manufacturers implement control strategies to maintain CPPs within acceptable ranges. Feedback control loops automatically adjust process parameters based on real-time measurements, while statistical process control uses statistical tools to monitor process trends and detect potential issues early. Meanwhile, alarm systems alert manufacturers when parameters approach or exceed acceptable limits. These advanced technologies should be fully integrated into the quality management ecosystem to protect data integrity, continued process verification, and regulatory compliance.
In-process sample analysis is a crucial component of biopharmaceutical quality control in biopharmaceutical manufacturing. It ensures product consistency throughout production and involves various techniques to monitor and control CQAs and CPPs during manufacturing. Implementing these techniques and strategies can help biopharmaceutical manufacturers ensure consistent product quality throughout the manufacturing process. This approach satisfies regulatory requirements and contributes to improved efficiency, reduced costs, and enhanced product safety and efficacy.
In-Process Sample Analysis Techniques
Sample analysis verifies product consistency and employs several tools, including:
- Chromatography: HPLC and related methods monitor protein concentration, assess product purity, and detect and quantify impurities or aggregates. Chromatography techniques separate complex biomolecules for analysis, providing detailed information about product quality.
- Spectroscopy: Spectroscopic tools provide real-time product aggregation and fragmentation measurement, rapidly detecting process deviations.
- Bioassays: Bioassays detect unexpected changes in samples from different stages of the bioprocess and can be performed under GMP or non-GMP conditions, depending on the stage of development
- PAT: Advanced technologies measure critical quality attributes in real-time, e.g., in-line sensors that track parameters like pH, temperature, and dissolved oxygen and spectroscopic methods that monitor product concentration or impurities. PAT tools integrate with data management systems for real-time analysis and control.
Ensuring Product Consistency
Drug products must demonstrate consistency to be safe, effective, and compliant. Manufacturers use advanced tools to monitor and ensure consistency in their comprehensive quality control scheme.
- Continuous monitoring uses in-line analyzers to monitor product flow directly, implements multiple analyzers at different points in the processing stream, and provides ongoing data collection and analysis to detect deviations quickly.
- Statistical process control relies on tools such as control charts to identify out-of-specification conditions and multivariate statistical process monitoring techniques like principal component analysis to monitor process trends.
- Risk mitigation via continuous monitoring reduces the risk of off-specification products being released, promptly identifies and corrects process deviations, and maintains regulatory compliance and product quality.
- Process understanding and optimization entail collecting process monitoring data to enhance understanding, fine-tune process parameters in real time, and support continuous improvement initiatives.
QC And Finished Product Testing
QC procedures test the final product to validate its identity, purity, potency, sterility, and safety. Additionally, stability testing measures the product's quality over long-term periods.
Regardless of methodology, all QC tests must be performed according to predefined specifications that are part of the product's registration with regulatory agencies. Deviating from these specifications without proper approval would constitute a severe GMP violation.
Release Testing Requirements
Release testing refers to the series of tests performed before a product can be approved for release to the market. These tests verify that the product meets all predetermined specifications and quality standards.
Types Of QC Tests Conducted Before Product Release
- Identity testing confirms that the product is, in fact, what it claims to be. Techniques such as HPLC, mass spectrometry, or immunological assays detect and verify the specific molecular characteristics of the biopharmaceutical product.
- Purity testing assesses the level of impurities in the product, typically through chromatographic methods like HPLC or electrophoresis that separate and quantify different components in the product to detect process-related impurities, product-related impurities, or contaminants.
- Potency testing measures the product's biological activity via bioassays or cell-based assays that quantify the product's ability to produce the intended biological effect.
- Sterility testing verifies that the product is free from microbial contamination using methods outlined in pharmacopeias like the USP or European Pharmacopoeia. Typically, incubated samples of the product are tested under conditions that would allow for microbial growth if contaminants were present.
- Safety testing may include tests for endotoxins, host cell proteins, or other process-related impurities. For injectable products, pyrogen testing may be required to ensure that the product will not induce a fever response when administered.
- Physicochemical testing includes a range of tests for parameters such as pH, osmolality, and particulate matter to ensure that the product's physical and chemical properties meet the required specifications.
- Content/strength testing quantifies the amount of active ingredient in the product so that each dose contains the correct amount of the active pharmaceutical ingredient.
Stability Testing Programs
Stability testing determines a product's shelf life over extended periods and assesses how the product's quality changes under various environmental conditions. Typically, a combination of physical tests (such as appearance and particulate matter), chemical tests (assay and impurities), microbiological tests (sterility and endotoxin levels), and functional tests (potency and, for solid dosage forms, dissolution) is conducted.
Long-term Product Quality Assessments
- Real-time stability studies involve storing products in their recommended environmental conditions and testing them at predetermined intervals, such as zero, three, six, 12, and 24 months, to assess long-term stability under normal storage conditions accurately. These studies determine the product's shelf life and storage recommendations.
- Accelerated stability studies store products under stress conditions, such as higher temperature and humidity, to provide quicker estimates of long-term stability and predict potential degradation pathways. While they don't replace real-time studies, they can provide valuable early insights into product stability.
- Forced degradation studies expose products to extreme conditions like heat, light, oxidation, and pH extremes to identify degradation mechanisms and further understand the product's stability profile and potential risks.
- Photostability testing assesses the effect of light exposure on product quality. This is particularly important for products exposed to light during storage or administration, and their results inform packaging decisions and storage recommendations.
- In-use stability testing evaluates product stability after opening or reconstitution. These tests are crucial for multi-dose products or those requiring reconstitution before use, ensuring the product remains stable and effective throughout its intended use.
The Importance Of Continuous Quality Monitoring
Ongoing quality monitoring and trending analysis ensure continuous process verification, enable early detection of issues and facilitate trend identification. This approach supports continuous improvement, regulatory compliance, and risk management. By collecting and analyzing data over time, manufacturers can make informed decisions, optimize processes, and maintain consistent product quality throughout the product lifecycle. This proactive stance aligns with modern quality management principles, builds product safety and efficacy confidence, and allows data-driven decision-making. Ultimately, these practices are crucial for maintaining high-quality standards in biopharmaceutical production.
What Analytical Methods Are Used In Biopharmaceutical QC?
Biopharmaceutical manufacturers employ advanced analytical techniques to thoroughly characterize their products, ensuring consistent quality, safety, and efficacy. A comprehensive approach to physicochemical analysis is essential for meeting regulatory requirements and maintaining the high standards expected in biopharmaceutical production.
Overview Of Physicochemical Analysis
Physicochemical analysis examines the physical and chemical properties of biological products to ensure their identity, purity, potency, and stability using the following techniques:
High-Performance Liquid Chromatography
HPLC is a versatile, widely used, and powerful analytical technique that separates, identifies, and quantifies components in a mixture by creating:
- Separation of proteins and peptides based on their physicochemical properties
- Quantification of active ingredients and impurities
- Assessment of product purity and heterogeneity
- Monitoring of post-translational modifications
HPLC can be used in various modes, such as reverse phase, size exclusion, and ion exchange, providing different information about the biopharmaceutical product. For instance, size exclusion chromatography is commonly used to detect and quantify protein aggregates, while reverse-phase HPLC can separate and quantify different protein variants.
Mass Spectrometry
Mass spectrometry (MS) is a powerful analytical tool that provides detailed information about the molecular structure and composition of biopharmaceuticals and is used for:
- Determining the exact molecular weight
- Identifying and characterizing post-translational modifications
- Sequencing proteins and peptides
- Detecting and quantifying impurities.
MS is often coupled with HPLC to provide separation and detailed structural information.
Electrophoresis Techniques
Electrophoresis techniques, such as gel electrophoresis and capillary electrophoresis, also separate proteins based on their size, charge, or both, as in:
- Sodium dodecyl sulfate-polyacrylamide gel electrophoresis for protein size analysis
- Isoelectric focusing for charge-based separation
- Capillary electrophoresis for high-resolution separation of proteins and peptides
Electrophoresis is often combined with MS. For example, proteins separated by gel electrophoresis can be excised and analyzed by MS for identification or further characterization.
Types Of Biological Assays
Biological assays play a crucial role in QA and QC by measuring the biological activity, potency, and other CQAs. Manufacturers frequently combine cell-based potency assays, enzyme-linked immunosorbent assay (ELISA), and flow cytometry to gain critical data that affects decision-making throughout the product lifecycle, from development through commercial production.
Cell-Based Potency Assays
Cell-based potency assays are designed to mimic the product's mechanism of action in the body, providing a functional measure of potency, and include:
- Mechanism of action: These assays reflect the product's intended biological effect. For example, a cell proliferation assay can be used for growth factors, while a cell death assay could be used for certain antibodies.
- Quantitative measurement: They provide a quantitative measure of potency, often expressed as relative to a reference standard.
- Variability management: Due to biological systems' inherent variability, these assays require careful design and statistical analysis to ensure reliability and reproducibility.
- Regulatory compliance: Regulatory agencies often require cell-based assays to demonstrate product potency throughout the product lifecycle.
- Lifecycle management: These assays need to be continuously monitored and potentially optimized over time to ensure they remain relevant and accurate.
ELISA
ELISA is commonly used to quantify specific proteins or antibodies in a sample and detects and quantifies process-related impurities, such as host cell proteins. For antibody-based therapeutics, ELISA can measure binding activity to specific antigens. Also, ELISA is amenable to high-throughput screening, making it suitable for routine QC testing.
Flow Cytometry
Both ELISA and flow cytometry complement cell-based assays by providing detailed molecular and cellular characterization of biopharmaceutical products. In particular, flow cytometry analyzes heterogeneous cell populations and the expression of specific proteins on cell surfaces while detecting impurities. Flow cytometry can also assess binding to specific cell types for antibody-based therapeutics.
How Microbiology Testing Informs QA/QC
Microbiology testing detects and controls microbial contamination, which is vital for QA and QC. Sterility testing, endotoxin testing, and bioburden analysis are commonly used methodologies.
Sterility Testing
Sterility testing, typically performed on the final product, ensures that pharmaceutical products are free from viable microorganisms, typically via membrane filtration and direct inoculation.
Two media types are typically used: fluid thioglycollate medium for anaerobic bacteria and soybean-casein digest medium for aerobic bacteria and fungi. Samples are usually incubated for 14 days, with regular checks for microbial growth.
Meanwhile, new technologies, such as adenosine triphosphate (ATP) bioluminescence and polymerase chain reaction-based methods, are being developed to reduce the time required for sterility testing. Closed-system sterility testing devices may provide recurrent sterile sample extraction for cell therapy products with a short shelf life.
Endotoxin Testing
Endotoxins, components of gram-negative bacterial cell walls that can cause severe patient reactions, must not be detected in biopharmaceutical products. The limulus amebocyte lysate (LAL) test, which uses the blood of horseshoe crabs to detect endotoxins, is susceptible and can detect endotoxins at deficient levels.
However, newer methodologies, such as kinetic chromogenic and turbidimetric assays, can provide faster results than traditional gel-clot methods. Another alternative to LAL, recombinant Factor C assays, can reduce reliance on horseshoe crab blood.
Bioburden Analysis
Bioburden analysis quantifies the number of viable microorganisms present in or on a product, component, or raw material before sterilization. Common methods include membrane filtration, pour plate, and spread plate techniques. Proper sampling techniques are crucial to ensure representative results.
These tests enumerate microorganisms and allow for trend analysis to identify potential sources of contamination in the manufacturing process. Bioburden data is used in risk assessments for sterilization processes and overall product quality.
Integration Into QA/QC Systems
Each of these microbiology tests is integrated into broader QA/QC systems. First, they complement environmental monitoring programs to ensure control of the manufacturing environment. Results are typically managed through laboratory information management Systems (LIMS) to safeguard data integrity and facilitate trend analysis.
Data from these tests drive continuous improvement in manufacturing processes and contamination control strategies. These tests are essential for meeting regulatory requirements that protect product safety and quality.
What Are the Best Practices For Quality Assurance Systems?
Biopharmaceutical companies should establish a robust quality culture that permeates all levels of the organization to meet regulatory requirements and best practices. A strong quality culture includes committed leadership, well-trained staff, QbD approaches, facility and equipment management, risk management, data integrity, CAPA systems, and strives for continuous improvement.
How To Establish A Robust Quality Culture
Biopharmaceutical manufacturing directly impacts human health and must thus meet high-quality standards. Focusing on personnel is the first step in establishing a robust quality culture.
What Is Leadership’s Role In Quality Management?
Establishing and maintaining high-quality standards begins at the top. Senior management must make quality a core value of the organization. This sets the tone for the company and prioritizes resources for quality initiatives, including technology, training, and personnel.
Leaders must hold themselves and others accountable for quality outcomes throughout the manufacturing lifecycle while encouraging continuous improvement and innovation. Clear, consistent communication about quality expectations and achievements is paramount, and quality considerations should be baked into all strategic decisions.
How Do Staff Training And Certification Maintain QA/QC Standards?
Staff must receive appropriate training and certification to meet high-quality expectations. Comprehensive QA/QC training ensures that staff has the required skills and knowledge to fulfill their roles in the quality ecosystem and meet regulatory standards.
When thoroughly trained, staff are less likely to make costly mistakes and can adapt more easily to new technologies and processes in the emerging Pharma 4.0 landscape. Training programs should focus on technical skills and foster a mindset prioritizing quality at every level.
Industry-recognized certifications further validate staff competencies and enhance the organization's credibility. By investing in robust training and certification programs, companies can ensure their workforce is well-equipped to maintain the highest quality standards.
What Are the Best Practices For Ongoing Personnel Qualification?
Maintaining and updating personnel qualifications is an ongoing process that requires a multifaceted approach. Staff should be regularly assessed to identify areas of improvement and given opportunities for continuing education to stay up to date on industry developments. Cross-functional training enhances flexibility and understanding of the entire quality process, while mentorship programs encourage experienced staff to guide and train newer employees, facilitating knowledge transfer.
Developing and tracking performance metrics related to quality can help identify specific training needs, and leveraging digital tools and platforms for training and qualification tracking streamlines this process. Staff should also be encouraged to give feedback on training programs and suggest improvements. Finally, qualification programs should be regularly updated to reflect changes in regulatory requirements, ensuring that the workforce remains current with industry best practices.
Why QbD Is Essential To QA/QC Management
Utilizing QbD principles creates an integrated, risk-based, and scientifically grounded approach to QA/QC. QbD emphasizes a proactive, holistic strategy that builds quality into the entire development and manufacturing process.
QbD’s risk-based approach assesses and manages risks throughout the product lifecycle, freeing up QA/QC teams to focus on the most critical aspects of product quality. This approach can reduce the overall testing burden while improving outcomes.
Targeted testing strategies based on CPPs and CMAs reflect a deep understanding of the process and product. Also, QbD encourages using a design space, a multidimensional combination of input variables, and process parameters that ensure consistent product quality. This gives QA/QC teams a framework for assessing whether a process is in control and producing quality products.
Additionally, QbD emphasizes monitoring critical parameters in real-time to make immediate adjustments and reduce the reliance on end-product testing, streamlining QC processes and enabling continuous improvement. By designing quality into the product and process from the beginning, QbD provides consistency and reliability in product quality and regulatory compliance.
How Quality Management Evaluates Facilities And Equipment
Biomanufacturing facilities and equipment must meet high industry standards to meet stringent quality standards. A strategy that includes preventive maintenance, environmental monitoring, cleanroom standards, equipment lifecycle management, and data-driven performance analysis ensures operational efficiency, safety, and regulatory compliance. A comprehensive QA/QC approach prevents contamination, minimizes downtime, extends equipment lifespan, and supports overall product quality.
Environmental Monitoring And The Importance of Cleanroom Standards
Environmental monitoring, particularly the management of cleanroom conditions, is a key component in a holistic quality strategy. Cleanrooms minimize contamination risks while complying with regulatory guidelines.
Standards like ISO 14644 and EU GMP Annex 1 provide guidelines for designing, operating, and monitoring cleanrooms. They specify requirements for air cleanliness, air flow, pressure differentials, and other environmental parameters. These standards establish a consistent framework for contamination control, provide measurable criteria for cleanroom performance, support regulatory compliance, and facilitate audits.
Cleanrooms are typically classified based on the maximum allowable number of particles per cubic meter of air. For example, an ISO 5 / Grade A cleanroom, often used for aseptic processing, has very stringent particle limits to ensure the highest level of cleanliness.
Monitoring Microbial And Particulate Contamination
Environmental monitoring programs in cleanrooms focus on both microbial and particulate contamination. Periodic sampling assesses the cleanliness of the cleanroom's air, surfaces, and personnel.
Active air sampling uses devices that draw in a specific air volume and capture particles or microorganisms on a collection medium. Passive air sampling exposes settle plates containing growth media to the cleanroom air for a set period. Continuous particle monitoring, however, relies on electronic devices to provide real-time data on airborne particle levels.
Surface monitoring often uses agar-filled plates pressed directly onto surfaces to collect microorganisms, while swabs can sample irregular surfaces or hard-to-reach areas. Rapid detection methods like ATP bioluminescence can provide quick results for surface cleanliness.
Personnel monitoring tests cleanroom staff for contaminants, including testing their gloves and garments for microbial contamination.
Based on a risk assessment, cleanroom monitoring methods are typically employed at defined frequencies and locations. The data collected is then analyzed to identify trends, investigate excursions, and drive continuous improvement in contamination control practices.
Equipment Validation And Commissioning
Equipment validation is required by regulatory bodies, while commissioning focuses on engineering and operational aspects. Together, they establish a solid foundation for consistent, high-quality biopharmaceutical manufacturing.
Validation includes qualification, demonstrates and documents that equipment and systems consistently perform as intended for their specific use, particularly regarding product quality and patient safety, and includes:
- Installation qualification: Verifies proper installation and configuration
- Operational qualification: Confirms operation within defined parameters
- Performance qualification: Demonstrates consistent performance under real-world conditions
Commissioning is the systematic process of verifying that equipment and systems are correctly designed, installed, and functioning according to operational requirements. It involves design reviews, installation checks, and functional testing.
Common Cleaning Validation Methods
Cleaning validation systematically demonstrates that cleaning procedures consistently and effectively remove residues to predetermined levels. Regulatory bodies like the FDA expect companies to have modern, accessible systems to maintain these records, ensure data integrity, and consistently adhere to validated cleaning processes. If an audit or inspection occurs, companies are expected to present this documentation as evidence of compliance. Techniques for verifying equipment cleanliness include:
- Visual inspection is the first step in assessing cleanliness. While not sufficient on its own, it can detect gross contamination.
- Swab sampling involves physically swabbing equipment surfaces to collect residues for analysis. It's particularly useful for hard-to-reach areas and provides direct evidence of surface cleanliness.
- Rinse sampling collects and analyzes the final rinse solution from the cleaning processes. It's effective for large surface areas but may not detect localized contamination.
- ATP bioluminescence rapidly detects the presence of ATP, indicating biological residues, but it doesn't identify specific contaminants.
- Specific analytical methods like HPLC, spectrophotometry, or TOC analysis detect and quantify specific residues or cleaning agents.
- Placebo batch testing through cleaned equipment tests for carryovers can provide comprehensive evidence of cleanliness.
Documentation And Regulatory Compliance
Regulatory agencies require documentation of cleaning procedures and must include the following:
- Cleaning protocols for equipment, including cleaning agents and methods
- Sampling plans, e.g., locations, methods, and frequencies for each equipment or facility area
- Analytical methods including specificity, sensitivity, and acceptance criteria
- Cleaning validation reports for the entire validation process, including results, data analysis, and conclusions
- Change control records of any changes to cleaning processes, equipment, or products that may impact cleaning effectiveness
- Training records to show that personnel are properly trained in cleaning and validation procedures
- Periodic review documentation to ensure ongoing effectiveness
- Deviation reports on any deviations from established procedures and subsequent corrective actions
What Are Risk Management And Root Cause Analysis?
Risk management and root cause analysis (RCA) work together to identify, analyze, and mitigate potential issues before they occur or prevent them from recurring. Risk management systematically identifies, assesses, and prioritizes risks, followed by coordinated efforts to minimize, monitor, and control their probability and impact. Conversely, RCA is a problem-solving method to identify the underlying causes of problems or events rather than just addressing their symptoms. Together, they enable organizations to proactively manage risks and systematically solve problems when they do occur.
Common Tools For Risk Assessment
Failure Mode And Effects Analysis (FMEA)
FMEA is a systematic, proactive method that identifies where and how a given process might fail and assesses the relative impact of different failures. It prioritizes risks based on their severity, occurrence, and detectability by:
- Identifying potential failure modes
- Determining their effects
- Assessing their severity, occurrence, and detectability
- Calculating the risk priority number
- Developing and implementing corrective actions
Fishbone Diagrams (aka Ishikawa Diagrams)
Fishbone diagrams are visual tools used to identify potential causes of a problem. The term "fishbone" refers to the shape of the final diagram, which resembles a fish skeleton. The primary categories of potential causes are typically represented as the main "bones," with more specific causes branching off from these. Common categories in a fishbone diagram include:
- People
- Methods
- Machines
- Materials
- Measurements
- Environment
This tool helps teams brainstorm and visualize all potential causes of a problem, ensuring a comprehensive analysis.
How Root Cause Analysis Enables Continuous Improvement
RCA involves several key steps. First, the problem must be clearly defined and understood by gathering all relevant information about the issue, including when and where it occurred, who was involved, and its immediate impact. Next, potential causes are identified through brainstorming sessions or structured tools like fishbone diagrams.
After identifying potential causes, the analysis moves deeper to find the root cause. This is where techniques like the 5 Whys come into play, encouraging analysts to keep asking "why" until they reach the fundamental reason for the problem. It's important to note that multiple root causes for a single issue may exist, and all should be explored.
RCA aims to identify the root cause and develop and implement effective solutions that address the fundamental issue rather than just its symptoms. These solutions should be implemented carefully, with clear plans for monitoring their effectiveness.
RCA is most effective when integrated into an organization's culture of continuous quality improvement to identify potential issues before they become serious problems and reactively address issues that have already occurred.
Protecting Data Integrity And Electronic Records Management
Data integrity and electronic records management are key components of QA/QC systems as they ensure data accuracy, consistency, and reliability throughout the product lifecycle. Key aspects include:
ALCOA+ Principles
ALCOA principles are adopted across the industry to enshrine data integrity, and the + adds Complete, Consistent, Enduring, and Available to the original acronym. These principles guide the creation, processing, and maintenance of bioprocessing data.
Electronic Records Systems
Electronic records systems are necessary in today's data-heavy industry. Still, they must undergo validation to prove they can reliably and securely create, modify, maintain, archive, retrieve, and transmit electronic records. LIMS, electronic batch records, and MES are all subject to regulations.
Audit Trails
Electronic systems must maintain secure, computer-generated, time-stamped, and readily available audit trails that record all data entries, modifications, and deletions.
Data Lifecycle Management
Data must be managed and controlled from its creation through processing, review, reporting, and archiving to eventual destruction. Each stage must maintain data integrity and comply with regulatory requirements.
System Validation
All computerized systems used in GxP (Good x Practice) environments must be validated to demonstrate their ability to consistently produce accurate and reliable data, including risk assessments, user requirement specifications, functional specifications, and testing protocols.
Data Backup and Recovery
Electronic records must be backed up and recoverable in case of system failures.
Access Controls and Data Security
Appropriate access controls, user authentication, and data encryption prevent unauthorized access or data manipulation.
Training and Procedures
Comprehensive training programs and SOPs for personnel creating, processing, and reviewing electronic records are vital.
Data Review and Approval Processes
Electronic data review and approval systems must maintain data integrity and comply with regulatory requirements for electronic signatures.
Cloud Systems and Contract Labs
As the use of cloud-based systems and contract laboratories increases, ensuring data integrity across these platforms and organizations becomes more complex but equally important.
Legacy Systems Integration
Many bioprocessing facilities still use legacy systems that may not have been designed with current data integrity standards in mind. Integrating these systems with modern platforms while maintaining data integrity is challenging.
Continuous Monitoring and Improvement
Continuous monitoring systems protect data integrity and steer processes towards ongoing improvement based on identified issues or new regulatory guidance as the industry evolves into Pharma 4.0.
CAPA’s Role In QA/QC Systems
CAPA takes a systematic approach to identifying, addressing, and preventing quality issues. A robust CAPA strategy addresses immediate quality issues while contributing to long-term quality improvement and compliance. CAPA is a powerful tool in the quality toolbox that drives continuous improvement and maintains high standards.
Role Of CAPA In Identifying And Correcting Deviations
CAPA is a structured approach to recognizing quality issues or potential problems from various sources. Once a deviation is identified, CAPA procedures guide the investigation to the root cause of the problem via a systematic analysis to understand why the deviation occurred.
Once the immediate issue is resolved, CAPA systems implement measures to prevent the problem or similar issues from reoccurring. Solutions may include process changes, additional controls, or staff training. By addressing both current and potential future issues, CAPA provides ongoing improvement of quality systems and processes.
Best Practices For CAPA Implementation And Monitoring
An effective CAPA system leverages several best practices:
- Clear procedures: Create well-defined, documented procedures for the CAPA process, e.g., problem identification, investigation, action planning, implementation, and effectiveness verification.
- Risk-based approach: Prioritize CAPA stratagems based on the identified issues’ risk and potential impact to optimize resource allocation.
- Thorough documentation: Maintain comprehensive records of all CAPA activities, including the initial problem description, root cause analysis, action plans, and verification results.
- Cross-functional involvement: Engage relevant stakeholders from different departments in the CAPA process for a holistic approach to problem-solving and solution implementation.
- Timely implementation: Set and adhere to realistic timelines for implementing corrective and preventive actions. Delays can lead to recurring problems or regulatory non-compliance.
- Effectiveness verification: After implementing CAPA, systematically verify and validate that the actions taken have effectively addressed the issue and prevented recurrence.
- Management review: Regularly review CAPA data and trends as part of management review processes to identify systemic issues and opportunities for improvement.
- Training and communication: Ensure that all relevant personnel are trained in CAPA procedures and communicate clearly regarding CAPA activities and outcomes to the entire organization.
- Continuous monitoring: Track processes and quality indicators to detect potential issues early and trigger preventive actions when necessary.
- Integrate with QMS: Thoroughly integrate the CAPA with other QMS elements, such as change control, document management, and training.
Why Continuous Improvement Is Crucial To QMS
Lean and Six Sigma principles are powerful methodologies that enhance the efficiency and effectiveness of quality management processes when applied to QA and QC.
Lean Principles In QA/QC
Lean’s systematic approach to reducing waste and maximizing values can be applied to QA and QC in the following steps:
- Identify value: Define the essential CQAs from the end-user's perspective.
- Map the value stream: Visualize the entire QA/QC process, from raw material inspection to final product testing, to identify non-value-adding activities or redundancies in quality processes.
- Create flow: Streamline quality processes to ensure smooth, continuous testing and inspection without unnecessary delays or bottlenecks.
- Establish pull: Implement just-in-time quality checks, ensuring quality inspections are performed only when needed, reducing over-processing and waiting times.
- Seek constant improvement: Continuously refine QA/QC processes, adopt new technologies, and update quality standards based on feedback and performance data.
Six Sigma Principles In QA/QC
Six Sigma principles rely on data to reduce variability and defects by using the following framework:
- Define: Outline quality objectives, patient requirements, and critical-to-quality characteristics.
- Measure: Implement robust measurement systems to assess product quality and process performance accurately.
- Analyze: Leverage statistical tools to identify root causes of quality issues and process variability.
- Improve: Develop and implement solutions to enhance quality and reduce defects.
- Control: Establish mechanisms to sustain improvements and prevent quality issues from recurring.
Integrating Lean And Six Sigma In QA/QC
When Lean and Six Sigma principles are integrated into a holistic QMS, they provide many benefits:
- Waste reduction: Lean principles identify and eliminate non-value-adding activities in quality processes, while Six Sigma tools reduce variability and defects.
- Data-driven decision making: Six Sigma's statistical approach complements Lean's focus on efficiency by providing quantitative insights for process improvement.
- Continuous improvement: Both methodologies emphasize ongoing refinement of processes, creating a culture of constant quality enhancement.
- Employee involvement: Lean and Six Sigma stress the importance of engaging all employees in quality efforts, fostering a quality-focused culture.
- Process standardization: Lean's emphasis on standardized work aligns with Six Sigma's goal of reducing variability, leading to more consistent quality outcomes.
- Root cause analysis: Six Sigma tools for root cause analysis complement Lean's problem-solving approaches, enabling more effective resolution of quality issues.
- Visual management: Lean principles of visual management enhance the transparency of quality metrics and performance, supporting Six Sigma's data-driven approach.
What Common Challenges Do Biopharmaceutical Companies Face When Implementing A QMS?
Managing QA/QC for biopharmaceuticals is challenging, given the complex nature of biologics, stringent regulatory requirements, heightened contamination risks, and the difficulties in producing them at scale.
Complexity Of Biologics Vs. Small Molecule Drugs
Biologics are more sensitive and complex than small molecule drugs and thus carry unique challenges and risks that must be mitigated with robust QA/QC procedures. A holistic QA/QC strategy must anticipate the following:
Structural Complexity
Small molecules typically have well-defined chemical structures, but biologics can have complex three-dimensional structures with post-translational modifications. Thus, QA/QC for biologics must account for a wider range of potential variations and impurities.
Manufacturing Process
Small molecule drugs are produced through chemical synthesis, which is relatively predictable and easy to control. In contrast, biologics are produced by living organisms or cells, necessitating a manufacturing process that is highly variable and sensitive to environmental conditions. Rigorous and comprehensive QA/QC measures are necessary to protect product quality throughout production.
Analytical Methods
Biologics' inherent complexities require sophisticated and diverse analytical methods for QA/QC. While small-molecule drugs can be characterized via straightforward chemical analyses, biologics require a combination of physicochemical, immunochemical, and biological assays to characterize the product thoroughly.
Stability And Storage
Small-molecule drugs are typically more stable and are often formulated for oral administration. Biologics, however, tend to be heat-sensitive and require special storage conditions, so QA/QC for biologics demands extensive stability testing and monitoring throughout the supply chain.
Immunogenicity
Biologics can elicit immune responses in patients, which is rare with small-molecule drugs. QA/QC initiatives for biologics must include testing for potential immunogenicity and monitoring for any changes that could affect the product's immunogenic profile.
Process Changes
Unlike small-molecule drugs, even small process changes can alter biologics' quality and efficacy. QA/QA for biologics must vigilantly monitor and control all aspects of the manufacturing process to avoid product deviations.
Regulatory Requirements
Due to their complexity, biologics are subject to stringent regulatory guidelines that require extensive, highly detailed QA/QC documentation and testing procedures.
Characterization Challenges
Characterizing a biological product is uniquely challenging compared to small molecule drugs. QA/QC initiatives must employ a wide range of analytical techniques and often rely on a combination of methods to ensure product quality and consistency.
Mitigating Supply Chain Risks And Raw Material Variability
Raw material availability and supply chain variability significantly impact biopharmaceutical product quality and manufacturing control. Even minor changes in raw material composition or properties can alter product identity, purity, potency, and stability. Inconsistencies also lead to process variability, affecting production yields and efficiency, making maintaining consistent product quality across batches challenging.
Increasingly, manufacturers outsource raw materials internationally, making consistency and traceability more complex. If there is a lack of transparency in the supply chain, it becomes more difficult to identify and control sources of variability and create scale-up challenges.
Variability in raw materials can introduce trace impurities or contaminants to the final product, altering a batch's efficacy and safety and potentially leading to batch failures. Due to the high number of raw materials involved in creating biologics, finding the root cause of a problem can be challenging, making effective control strategies difficult to implement.
Raw material variability can also affect the analytical methods used for product characterization and release testing, leading to inconsistent or unreliable test results and further complicating quality control efforts. From a regulatory perspective, variability in raw materials can lead to out-of-specification (OOS) results for drug products, impacting patient safety, the drug product supply chain, and regulatory submissions.
To address these challenges, biopharmaceutical companies are implementing strategies like robust supplier qualification and auditing processes, comprehensive raw material characterization methods, and risk-based approaches to manage raw material variability. Companies are also enhancing supply chain transparency and traceability, investing in advanced analytics and PAT to monitor and control variability in real-time, and collaborating with suppliers to improve raw material consistency and quality.
Scaling From R&D To Commercial Production
Scaling biopharmaceutical production from the lab to commercial levels presents unique QA/QC challenges due to the complexity of biological processes and products. Maintaining consistent quality over increasing production volumes requires comprehensive, proactive QA/QC strategies.
First, the process must remain consistent across different scales. Methodologies that create reliable products for small-scale R&D may not work at larger volumes, potentially affecting CQAs. QA/QC teams must develop and validate scalable analytical methods that accurately characterize the product at each stage.
Sterile environments are more difficult to maintain as production scales up, requiring robust environmental monitoring programs and sterilization processes suitable for large-scale operations.
Data management and integrity present are another scale-up challenge. As process and analytical data increase, QA/QC systems must be in place to guarantee data accuracy, traceability, and compliance with regulatory requirements.
If outsourcing to a CMO, tech transfer introduces additional QA/QC challenges. Ensuring the process performs consistently in a different facility requires thorough documentation, clear communication, and rigorous testing to verify equivalence.
Throughout scale-up, QA/QC teams must continually assess and mitigate risks to product quality by establishing in-process controls, implementing real-time monitoring systems, and enforcing effective CAPA processes.
Solutions For Overcoming Compliance And Regulatory Hurdles
As innovative biopharmaceutical products advance and become more prevalent, the regulatory environment has adapted to these new therapeutics, creating stringent requirements to protect patient safety. However, biopharmaceutical companies can navigate this byzantine landscape, reduce compliance risks, and improve operations as part of a comprehensive QA/QC strategy.
Product Complexity And Variability
Complicated biopharmaceutical products are highly sensitive to variations but must be manufactured consistently to meet regulatory approval and protect patient health. To address these difficulties, companies should:
- Implement robust process control measures and real-time monitoring systems
- Adopt QbD principles to build quality into products from the early stages of development
- Invest in advanced analytical techniques to better characterize and control product variability
Evolving Regulatory Requirements
In response to the evolving therapeutic landscape, regulatory agencies frequently update their guidelines and requirements, making it challenging for companies to stay compliant. However, companies can take proactive measures, such as:
- Engage regulatory intelligence teams to stay informed about changes.
- Create flexible QMSs that can adapt to new requirements.
- Participate in industry collaborations and regulatory harmonization efforts.
GMP Compliance
Complying with GMP standards requires strict quality control at all stages of production, and companies should:
- Design comprehensive QMSs
- Conduct regular internal audits and mock inspections
- Provide employee training and development programs focused on GMP compliance
Data Integrity And Management
Ensuring the accuracy, security, and traceability of data throughout the manufacturing process can be achieved with these tools:
- Robust digital systems for data collection, analysis, and management
- Electronic batch record systems
- Enhanced cybersecurity measures that protect sensitive data
Supply Chain Oversight
Regulatory bodies expect rigorous oversight of the entire supply chain, which can be complex for global companies. To mitigate supply chain risks, companies should:
- Require comprehensive supplier qualification and auditing programs
- Leverage track-and-trace technologies to ensure product integrity throughout the supply chain
- Develop risk-based approaches to supplier management
Laboratory Controls
Establishing scientifically sound and appropriate specifications, standards, and test procedures is crucial for ensuring product quality. Companies can:
- Invest in state-of-the-art analytical equipment and methodologies
- Apply robust method validation processes
- Enhance LIMS to improve data management and traceability
Post-Marketing Surveillance
Continuous monitoring of product performance and safety after market approval is a regulatory requirement that can be resource-intensive, so innovative companies rely on the following:
- Advanced pharmacovigilance systems
- Real-world data and analytics that monitor product performance
- Proactive risk management strategies for marketed products
Managing Deviations And Nonconformities
Deviations and nonconformities are related but distinct hurdles in bioprocessing. Deviations are any departure from an approved instruction, procedure, established standard, or specification and typically occur during the manufacturing process or testing procedures. However, nonconformities (also called nonconformances) are instances where products or components do not meet specified standards or requirements.
Both deviations and non-conformities have severe implications for product quality and regulatory compliance. For instance, a temperature deviation during cell culture could lead to changes in protein glycosylation patterns, potentially affecting the product's biological activity. An OOS due to a nonconformity could lead to batch rejection, causing supply chain disruptions and financial losses.
By effectively managing deviations and non-conformities, bioprocessing companies can maintain high product quality standards, ensure patient safety, and meet regulatory requirements. A proactive QA/QC approach addresses immediate quality issues and contributes to continuous improvement in manufacturing processes and quality systems.
Bioprocessing Deviation Examples
- Temperature excursion during cell culture: If the bioreactor temperature deviates from the specified range, it could affect cell growth and product quality.
- pH fluctuation in buffer preparation: A deviation in pH during buffer preparation could impact downstream purification processes.
- Incorrect reagent concentration: Using a reagent at the wrong concentration during a manufacturing step would be considered a deviation.
- Deviation from SOP: For instance, if an operator skips a step in the cleaning procedure for equipment.
Examples Of Nonconformities
- OOS test results: If the final product fails to meet purity or potency specifications during quality control testing.
- Particulate matter in the final product: The presence of visible particles in a parenteral product would be a non-conformity.
- Incorrect labeling: If the product label contains erroneous information or lacks required details.
Best Practices For Responding To Deviations And Non-Conformities
- Prompt identification and reporting: Establish systems to detect and report deviations and non-conformities quickly.
- Thorough investigation: Conduct a comprehensive RCA to understand why the event occurred.
- Impact assessment: Evaluate the potential impact on product quality, patient safety, and regulatory compliance.
- CAPA: Implement measures to correct the immediate issue and prevent recurrence.
- Documentation: Maintain detailed records of the event, investigation, and actions.
- Risk-based approach: Classify deviations and non-conformities based on potential risk to prioritize response efforts.
- Trend analysis: Regularly review data to identify patterns or recurring issues that may indicate systemic problems.
- Training: Ensure personnel are well-trained to identify, report, and respond to deviations and non-conformities.
- Quality system improvement: Use insights from deviations and non-conformities to improve QMSs continually.
- Regulatory compliance: Ensure all actions comply with relevant regulations and guidelines, such as GMP requirements.
What Emerging Trends Are Revolutionizing Biopharmaceutical QA/QC?
As the industry evolves into Pharma 4.0, advanced technologies like AI and ML are taking center stage, poised to usher in an era of faster, higher-volume bioprocessing with fewer errors and less human intervention. Adopting these trends requires significant up-front investment, but it is increasingly necessary to remain competitive in the space.
Modernizing QA With Real-Time Release Testing (RTRT)
RTRT is an emerging methodology that radically changes how product quality is assured. Leveraging PAT and advanced control strategies, RTRT monitors CQAs in real-time during manufacturing, rather than relying on end-product testing to evaluate and assure product quality. This approach reflects a deep understanding of the manufacturing process, including the intricate relationships between process parameters and product attributes.
Implementing RTRT offers several advantages. Products can be released faster, facilities enjoy improved productivity, and costs may be decreased without compromising quality. However, successfully adopting RTRT requires a significant investment in advanced sensor technologies, sophisticated data analytics capabilities, and process control strategies.
Regulatory bodies are creating guidance to support RTRT's adoption, including ICH guidelines Q8, Q9, Q10, and Q11, which support the implementation of RTRT as part of a broader QbD approach.
How Continuous Bioprocessing And PAT Affect QA/QC
With Pharma 4.0 on the horizon, manufacturers are leveraging PAT systems to shift from batch to continuous manufacturing, which affects QA and QC strategies. First, continuous bioprocessing uses a QbD and control paradigm rather than the traditional quality-by-testing methodology thanks to real-time monitoring and dynamic quality management.
Deviations are immediately detected and corrected, reducing the risk of producing OOS products. A continuous data stream enables predictive models that anticipate potential quality issues, shifting QA/QC from a reactive to a proactive exercise.
Implementing continuous manufacturing and PAT can lead to faster release times and decreased inventory holding costs. However, it requires developing and validating stringent in-process testing methods that correlate with final product quality. Additionally, in continuous manufacturing, the concept of a "batch" becomes less clear, necessitating new strategies for product traceability and recall procedures.
These changes also demand new skills from QA/QC personnel, particularly in data analysis, statistical process control, and PAT tools. Regulatory compliance in this new paradigm depends upon thorough validation and documentation of these novel approaches.
The Role Of Automation, AI, and ML In QA/QC
Automation and AI are transforming traditional QC approaches to ensure product safety, efficacy, and regulatory compliance. Integrating these technologies enables more efficient, accurate, and proactive quality management strategies.
Automation And AI
In bioprocessing, automation and AI are being applied to various aspects of QA/QC:
- Real-time monitoring and control: AI-powered systems can continuously monitor critical process parameters and product quality attributes, enabling immediate detection and correction of deviations and reducing the risk of producing OOS products. For example, AI algorithms can analyze data from in-line sensors to detect subtle changes in cell culture conditions that might affect product quality.
- Automated visual inspection: AI-powered computer vision systems can perform high-speed, accurate assessments of final products, detecting defects that human inspectors might miss. This is particularly useful for detecting particulates, checking fill levels, or inspecting packaging integrity.
- Data integration and analysis: AI can integrate and analyze data from multiple sources across the bioprocessing workflow, providing a holistic view of product quality. This enables more informed decision-making and quickly identifies the root causes of quality issues.
Machine Learning
- Predictive analytics: Machine learning models analyze historical and real-time data to predict potential quality issues before they occur. For instance, predictive models forecast when equipment maintenance is needed to prevent contamination or process disruptions.
- Process optimization: Machine learning algorithms analyze complex datasets to identify optimal process parameters, potentially improving product quality and consistency while reducing waste.
- Pattern recognition: Machine learning algorithms identify complex patterns in process data that may indicate quality issues, even when these patterns are not obvious to human observers.
- Anomaly detection: Unsupervised learning algorithms detect anomalies in process data or product characteristics, flagging potential quality issues for further investigation.
- Process understanding: By analyzing large datasets, machine learning uncovers hidden relationships between process parameters and product quality, deepening our understanding of complex bioprocesses.
- Continuous improvement: Machine learning models learn from new data, improving their predictive accuracy and adapting to process changes.
How Digital Twins Enhance QMS
Digital twins enhance QA and QC measures by providing a comprehensive, real-time virtual representation of the production process. These advanced simulation models integrate data from various sources, including PAT, quality data, and time-series data, to create a dynamic replica of the manufacturing environment. By leveraging this technology, manufacturers monitor CQAs and KPIs in real-time to detect and correct deviations immediately.
Digital twins support process validation by establishing superior control models, reducing the number of process performance qualification (PPQ) runs, and facilitating continuous process improvement. They enable predictive analytics to anticipate and prevent quality issues before they occur. In bioprocessing, digital twins can simulate complex cell culture conditions, optimize upstream and downstream activities, and support batch-to-batch comparisons without disrupting ongoing processes.
Furthermore, they enhance cybersecurity by detecting potential intrusions while manufacturing continues. By providing a platform for testing and simulating various scenarios without risking actual production, digital twins enable efficient problem-solving and process optimization. Ultimately, this technology improves product quality, consistency, and regulatory compliance while potentially reducing development costs and time-to-market for new biopharmaceutical products.
Single-Use Technologies And Quality Management
The popularity of single-use systems (SUS), including disposable bioreactors, affects QA and QC processes. SUS reduces the risk of cross-contamination and minimizes human error by eliminating the need for cleaning, sterilization, and validation between batches. This shift allows QA/QC teams to focus on CQAs instead of time-consuming cleaning validation processes.
In particular, pre-sterilized disposable bioprocess bags are gaining popularity because they decrease sterilization time and costs. Disposable bioreactors are ideal for continuous manufacturing, improving scalability, efficiency, and product quality.
However, implementing SUS introduces new QA/QC challenges, e.g., enforcing rigorous supplier qualification and managing extractables and leachables. QA/QC strategies must include thorough evaluations of SUS component integrity, focus on supply chain management, and develop new testing protocols specific to SUS technologies.
Conclusion
QA and QC ensure product safety, efficacy, and regulatory compliance. From raw material testing to final product release, QA/QC processes safeguard the integrity of biopharmaceuticals throughout their lifecycle. As the biomanufacturing landscape evolves with advanced technologies and increasing regulatory scrutiny, QA/QC practices must adapt and innovate. Data integrity principles, risk-based approaches, and cutting-edge analytical methods are reshaping traditional QA/QC paradigms.
Companies must embrace robust, flexible QA/QC systems that leverage automation, real-time monitoring, and advanced data analytics to meet future challenges. By investing in comprehensive QA/QC strategies, organizations can ensure compliance and drive continuous improvement, enhance operational efficiency, and ultimately deliver safer, more effective treatments to patients.
Frequently Asked Questions (FAQs)
Frequently asked questions regarding QA and QC in bioprocessing include:
1. What is the difference between QA and QC in biomanufacturing?
While both QA and QC are essential for ensuring product quality, they have distinct roles:
QA:
- Focuses on preventing quality issues through systematic activities
- Develops and implements QMSs and procedures
- Conducts audits and oversees compliance with regulations
- Manages documentation and change control processes
- Provides training on quality-related topics
QC:
- Focuses on identifying quality issues through testing and inspection
- Performs analytical and microbiological testing of materials and products
- Monitors process parameters and product attributes
- Investigate deviations and out-of-specification results
- Maintains testing equipment and laboratory systems
Essentially, QA works to prevent quality problems, while QC works to detect them.
2. How does QA/QC contribute to regulatory compliance in biomanufacturing?
QA/QC plays a vital role in regulatory compliance by:
- Ensuring adherence to GMPs
- Maintaining accurate and complete documentation
- Supporting process validation activities
- Conducting internal audits and preparing for regulatory inspections
- Investigating and addressing deviations and non-conformances
- Implementing CAPA
- Ensuring data integrity in all quality-related activities
3. What is the role of QC in biopharma manufacturing?
QC in biopharmaceutical manufacturing is crucial in ensuring product safety, efficacy, and consistency. Key responsibilities include:
- Testing raw materials, in-process samples, and finished products
- Conducting analytical and microbiological testing
- Monitoring critical process parameters
- Identifying and investigating deviations or out-of-specification results
- Maintaining accurate documentation of all testing activities
- Supporting process validation and continuous improvement initiatives
QC helps verify that products meet predetermined specifications and comply with regulatory requirements before release.
4. What are some key quality control tests in bioprocessing?
Important QC tests in bioprocessing include:
- Protein concentration and purity assays (e.g., HPLC, electrophoresis)
- Bioactivity assays
- Endotoxin testing
- Sterility testing
- Viral clearance studies
- Cell-based potency assays
- Stability testing
- Particle size analysis
- Glycosylation profiling
5. What are some challenges in QA/QC for biopharmaceuticals?
Challenges of biopharmaceutical quality management include:
- Complexity and variability of biological products
- Rapid technological advancements requiring continuous training
- Increasing regulatory scrutiny and evolving guidelines
- Managing large volumes of data and ensuring data integrity
- Balancing speed-to-market pressures with rigorous quality standards
- Implementing effective QA/QC strategies for novel therapies (e.g., cell and gene therapies)
About The Author
Elizabeth Mann is a skilled writer with over a decade of experience in content creation, specializing in the life sciences industry. As a writer for Life Science Connect, she develops in-depth content that informs and engages professionals in the pharmaceutical, biotech, and medical device sectors. Her areas of focus include biologic drug production (including cell and gene therapies), clinical trial design and execution, and drug development and manufacturing outsourcing.
EXPERT INSIGHTS ON QA-QC
-
 Zero-Acceptance Sampling Can Protect Your Next Data Migration
Zero-Acceptance Sampling Can Protect Your Next Data MigrationWhen migrating data in a GMP setting, verifying every record is impossible. A well-structured sampling approach can mitigate the risks and help with a smooth transition.
-
 Why Aren't QC Labs Automated? Blame Sample Container Variability
Why Aren't QC Labs Automated? Blame Sample Container VariabilityMembers of the Laboratory Automation Plug and Play group speak up about the factors they see holding back quality control labs from achieving more robust automation.
-
 Count On Data Integrity Auditors Making These 10 Requests
Count On Data Integrity Auditors Making These 10 RequestsEvery audit, however unique, requires confirmation that effective data integrity governance exists, supported by strong SOPs, proper validation, and regulatory adherence.
-
 A Risk-Based Approach To Filter Integrity Testing Annex 1 Requirements For Biologics DS
A Risk-Based Approach To Filter Integrity Testing Annex 1 Requirements For Biologics DSHow do the principles of filter integrity testing described in EudraLex Annex 1 apply to low bioburden drug substance manufacturing? And how can using a risk-based approach help?
-
 A Comprehensive Guide For Supplier Quality Agreements
A Comprehensive Guide For Supplier Quality AgreementsThis overview of SQAs includes a handy matrix template, which is useful for determining who is responsible for managing specific responsibilities.





EDITORIAL PERSPECTIVES ON QA-QC
-
 Suppliers Pushing Novel Analytical Methods Testing Forward
Suppliers Pushing Novel Analytical Methods Testing ForwardThe urgency to develop novel therapeutics must be balanced by rigorous safety testing, but cell and gene therapy testing protocols, of which there are many, have to date been neither standard nor quick. What role are outsourced testing service providers playing to change that paradigm?
-
 Complex Protein Development: Assay Early, Assay Often
Complex Protein Development: Assay Early, Assay OftenExperts from FyoniBio, SOTIO, and Vera Therapeutics weigh in with first-hand experience on how process decisions made very early on can influence – beneficially or detrimentally – the efficiency of upstream and even downstream operations in the development of novel protein therapeutics.
-
 Considerations For AI Use In Biomanufacturing
Considerations For AI Use In BiomanufacturingExpert's from this month's Bioprocess Online Live event discuss different use cases for AI and shared their thoughts on the adoption of these technologies.
-
 Single Use In Biopharma: Beyond Savings & Sustainability
Single Use In Biopharma: Beyond Savings & SustainabilitySUT continues to trend in biopharmaceutical applications, driven largely by environmental and economic considerations. But there’s a lot more to the SUT story, including supply chain and standardization advantages. We dove headlong into those issues and more with independent SUT expert Paul Priebe and Krystal Biotech VP of Technical Operations Mark Petrich.
-
 Where's The Case For Generative AI In Biopharmaceutical Manufacturing?
Where's The Case For Generative AI In Biopharmaceutical Manufacturing?The early use cases for AI in the biopharmaceutical industry—at least, the early public use cases—have largely come from R&D, and more specifically, target identification and molecular design. Where are the use cases in biologics manufacturing, supply chain management, QMS, and operations, and what’s holding us back?





ON-DEMAND QA-QC WEBINARS
-
 Integrating CPV And APQR Data And Workflows To Reduce Redundant Activities
Integrating CPV And APQR Data And Workflows To Reduce Redundant ActivitiesExperts in the pharmaceutical industry share insights on how to effectively implement a strong, integrated CPV and APQR framework. Watch now and learn how automation enhances efficiency.
-
 Keeping Up With New Emerging Analytical Technology
Keeping Up With New Emerging Analytical TechnologyExplore advancements in emerging analytical technologies like Process Analytical Technology (PAT) and mass spectrometry, improving drug development through enhanced bioprocess monitoring and control.
-
 Analytical Solutions To Help Accelerate Bioprocessing Success
Analytical Solutions To Help Accelerate Bioprocessing SuccessUnlock the power of data-driven bioprocessing. Discover how advanced analytics can optimize your cell culture process, improve performance, and accelerate success.
-
 Navigating GxP Compliance Challenges
Navigating GxP Compliance ChallengesLearn how to select and manage trusted software suppliers to streamline your digital transformation, mitigate risks, and ensure regulatory adherence. Watch to discover best practices and real-world examples.
-
 Innovative Contamination Control: Enabling Integrity And Efficiency
Innovative Contamination Control: Enabling Integrity And EfficiencyAn integrated approach can enhance contamination control, boost process efficiency, and ensure the production of high-quality cell therapy products.




