
Computer Systems Validation Pitfalls, Part 2: Misinterpretations & Inefficiencies
By Allan Marinelli and Abhijit Menon

When work is not performed according to protocol instructions — by cutting corners on quality outputs with the focus on profit as the main driver or by creating unnecessary work that reduces efficiency — this can result in pharmaceutical, medical device, cell & gene therapy, and vaccine companies losing profits, efficiencies, and effectiveness. The reason is a lack of QA leadership and management/technical experience, among other poor management practices, coupled with the attitude that “Documentation is only documentation; get it done fast and let the regulatory inspectors catch us if they can find those quality deficiencies during Phase 1 of the capital project and our validation engineering clerks will correct them later”. Consequently, these attitudes can lead to at least 15 identified quality deficient performance output cases during the validation phases, as will be discussed in this four-part article series. See Part 1 here.1
Note that the QA rubber stamping approach has often been performed by numerous companies many decades ago and still in current times, since validation was often construed as being “not needed, or we will do this at the last minute to satisfy the regulators.” This was attested to by many business-driven upper management stakeholders across the board, despite being necessary to meet FDA and other regulatory bodies’ regulatory requirements when performing all testing, while also endorsing quality from the inception through the completion of any projects in order to substantiate that your processes, equipment, computerized systems, information systems, infrastructures, supply chain management, quality management systems, manufacturing/productions areas, etc., are consistently demonstrating a state of control.
In part 2 of this four-part series, we will examine the following quality deficiency performance output cases we have observed, including unnecessary “nice to haves” that waste execution efficiencies by not putting enough time where it matters:
- Not clearly defining the “family approach”
- Confusion interpreting calibration versus certified records and material being used to document in one attachment
- Previous QAs’ misinterpretation of protocols’ directives and proposing “nice to haves” that unnecessarily increase execution times (bottom-line: reducing the execution efficiencies and effectiveness)
- Misleading deviation identifications (numerous protocol identifications of the same deviation)
- Executions incomplete (status have not been completely reviewed and populated by the validation department) prior to submitting to QA
Not Clearly Defining The “Family Approach”
Company ABC used the family approach, which is defined as using the same or equivalently functional equipment/system for protocol execution while factoring in that the equipment/system would first require an initial full-fledged validation. (All of the identified intended functions to be used in manufacturing/production will be initially tested as part of the Operational/Functionality section of the protocol.) Subsequently, the next set of the same or equivalently functional equipment/system is executed by validating a partial functionality output of identified testing (only testing the critical portions of the functionalities) based on previously approving a functional risk assessment (as agreed upon by all applicable stakeholders).
Using the family approach, the same previously approved executable family of equipment/system protocols, accompanied by their respective attachments, will be used again, but at reduced testing (this would only require a subset of functional testing) of the Operational/Functional section of the protocol. This should inevitably increase the efficiency of execution, while maintaining the quality paradigms based on a previously approved risk assessment in alignment with the family approach.
However, if gaps are created by not clearly defining the family approach or by misconstruing the intended uses because of a lack of hands-on training relevant to the applicable stakeholders performing the validation executions, coupled with deficient QA technical management leadership, then the family approach becomes futile.
Example
The initial senior QA reviewing/approving authority involved in the capital project had an extensive background in project management with very little experience in QA or compliance throughout their 40+ years in the industry. This subsequently resulted in the approval of all the initial full-fledged validations for the various types of equipment/systems by adopting a QA rubber stamping approach, which upper management liked, since they wanted to have all validations completed within an unreasonably short period of time, regardless of whether quality was ever factored in.
As a consequence, the validation engineers, through no fault of their own, continued to repeat the same deficient validation execution habits by copying/pasting the execution references that were initially part of the initial full-fledged validations onto the next set of identical equipment/system for reduced testing during the subsequent execution phases.
This inevitably resulted in the initial senior QA reviewing/approving authority, who lacked QA experience, to work on other capital projects while continuing to be the initial senior QA reviewing/approving authority on those other projects without any hassles, based on the directives of upper management.
Confusion Interpreting Calibration Versus Certified Records And Material Being Used To Document In One Attachment
As part of documenting evidence in one attachment, the data must be entered in their respective column fields that specify whether the external equipment being used had existing calibration records that are calibration NIST traceable/equivalent. These would then inevitably require subsequent recalibration after the calibration due date, which would eventually expire, unless the main equipment/system to be validated had been classified as single-use and given a “one-time use calibrated” record from the manufacturer. Therefore, one needs to understand the differences between external equipment that is intended to be used to conduct respective measurements of the main piece of equipment/system to be validated versus the main equipment/system itself.
Below are three examples of inconsistencies that were previously approved by the initial senior QA (who lacked technical hands-on QA experience but had 40 years of project management experience) during the initial full-fledged validation. This shows the initial senior QA did not understand how to provide advice to the respective validation stakeholders so the validation stakeholders could adjust for corrections based on the intended uses of Attachment A, titled External Test Equipment and Materials, upon populating the identified column fields during the execution phase.
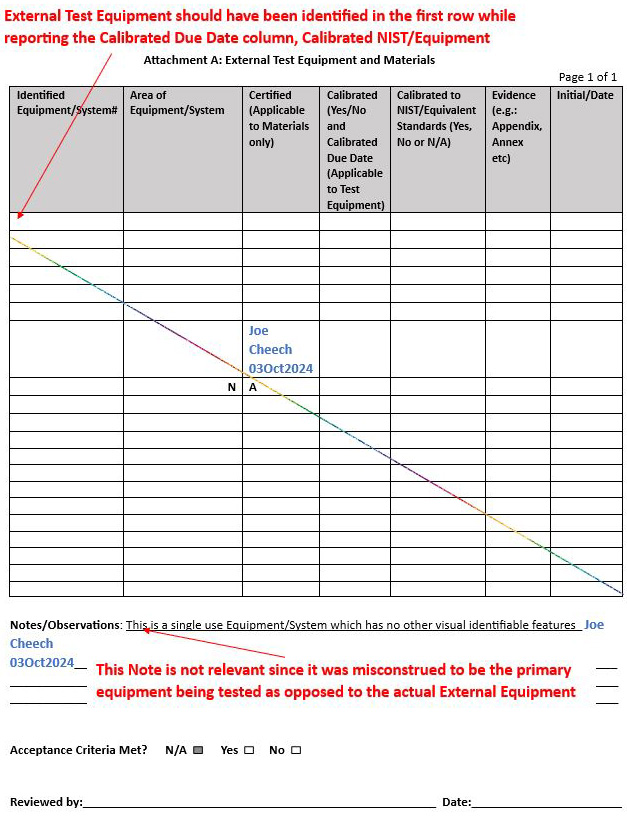
Example 1: Attachment A was completed using N/As as opposed to completing the required fields/columns
Solution: Since an external test equipment was used to measure the RPM of the centrifuge, the speed testing equipment (external equipment) should have been identified on Attachment A while populating the calibrated and calibrated NIST/equipment columns, accompanied by the respective supporting evidence (by attaching the current calibration certificate of the external test equipment), which would have minimally shown the calibration due date and the NIST/equivalent calibration methodology, among other attributes.
See the notes outlined in red by the technically experienced senior QA to represent the solution or clarification of intent below:
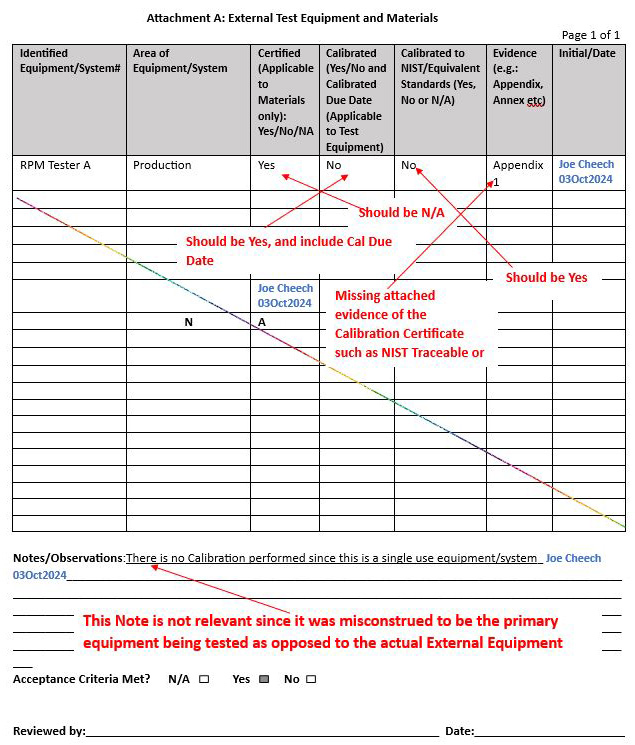
Example 2: Attachment A: Confusion interpreting external test equipment versus protocol main equipment/system
Solution: See the notes outlined in red by the technically experienced senior QA to represent the solution or clarification of intent below:
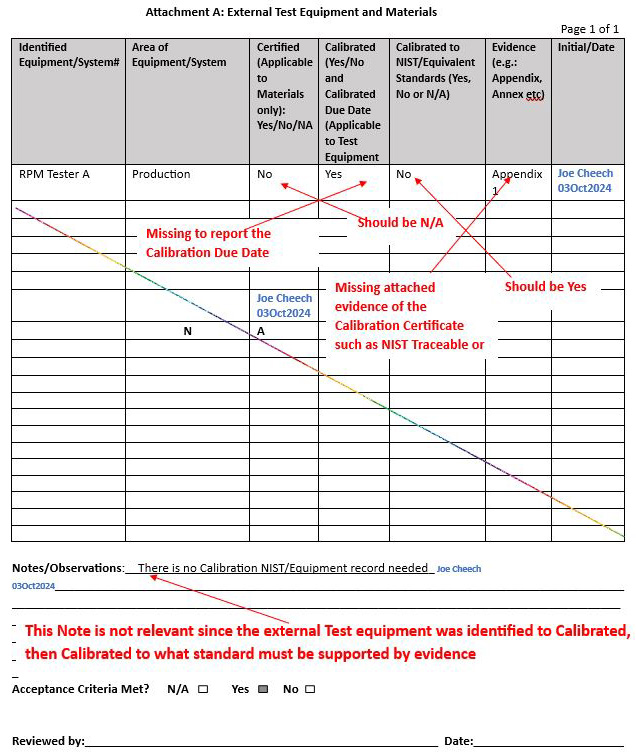
Example 3: Attachment A: Not understanding the information on a calibration record and not understanding the NIST traceability paradigm
Solution: See the notes outlined in red by the technically experienced senior QA to represent the solution or clarification of intent below:
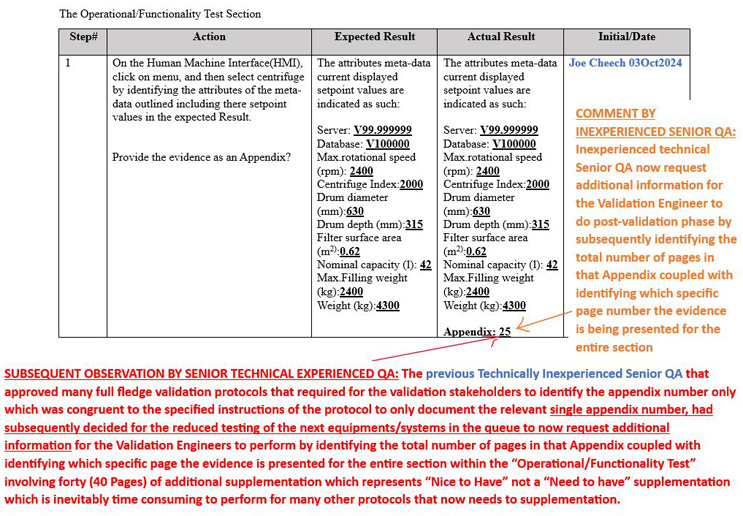
Previous QAs’ Misinterpretation Of Protocols’ Directives And Proposing “Nice To Haves” That Unnecessarily Increase Execution Times (Bottom-Line: Reducing Execution Efficiencies And Effectiveness)
While endorsing the continuous improvement paradigm with the aim of increasing efficiency/effectiveness can be looked upon as a good thing, it is important to ensure that all the instructions are performed as stipulated in the protocol without adding “nice to haves” over and above what has already been pre-approved for minimal performance.
Solution: See the notes outlined in red to represent the solution or clarification of intent by the technically experienced senior QA versus the comments shown in orange made by an inexperienced senior QA.
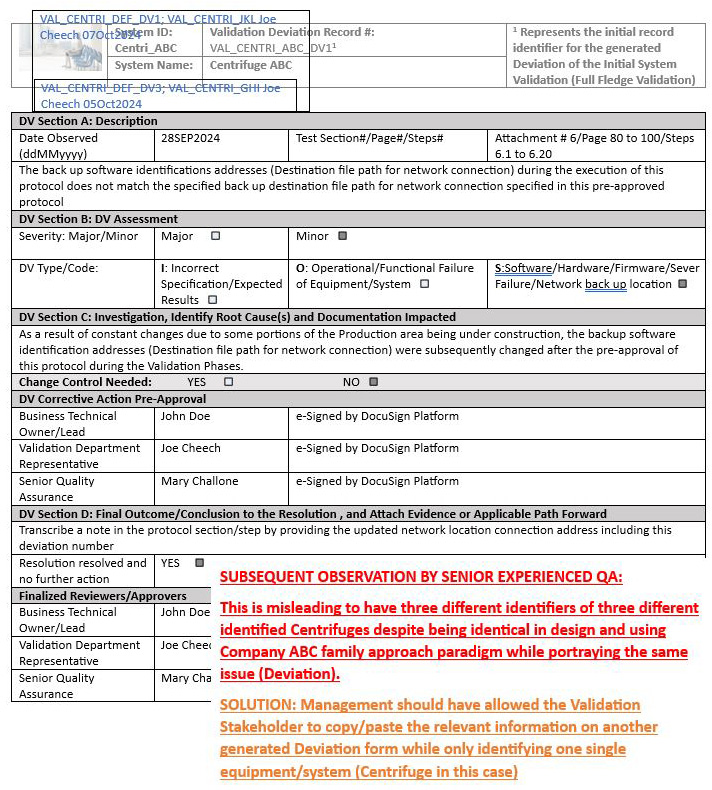
Misleading Deviation Identifications (Numerous Protocol Identifications On The Same Deviation)
The initial protocol pertaining to Centrifuge ABC required initial full-fledged validation while any other subsequent validation executions involving the same identical equipment/system by design will only require a partial validation of the operational/functional test section. This is based on a previously approved risk assessment that was put together in alignment with the family approach that was defined in the Validation Master Plan, whereby only the critical identified operational/functional test shall be repeated for the next identical set of equipment/system.
Therefore, this led to three different identified labels for a single deviation discrepancy, which can be seen below. The initial centrifuge identified within the center of the header is shown in black, which represented the initial full-fledged validations for Centrifuge ABC, while the other two centrifuges, Centrifuge GHI and Centrifuge JKL, are identified in blue.
Solution: See the notes outlined by the experienced senior QA reviewer/approver in red to stipulate the reasoning of the misleading deviation identification issue followed by a proposal transcribed in orange below.
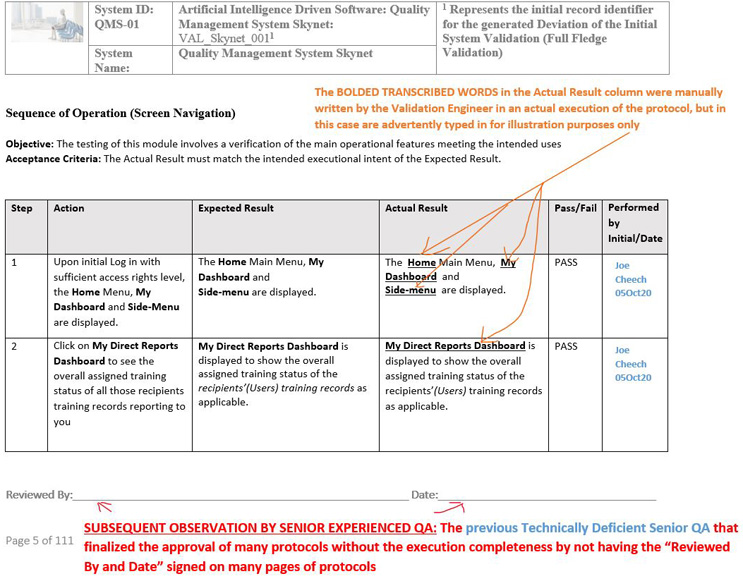
Executions Are Incomplete (Not Reviewed And Populated By The Validation Department) Prior To Submitting To QA
The QA department should always receive a fully executed original version of the completed protocol including all respective wet signatures (handwritten initial and date) for each of the relevant steps, with attachments, coupled with the review/approval by wet signatures/dates or e-signatures if using an electronic methodology (by first generating a “review workflow” using the electronic validated quality management system platform followed by “an approval workflow” after all of the comments provided by the involved stakeholders have been addressed) of all of the applicable stakeholders. This must be performed in alignment with the instructions of the protocol prior to submitting the execution of the protocol for QA final approval.
Solution: See the notes outlined in red by the technically experienced senior QA to represent the solution or clarification of intent below:
Conclusion
Ensure that the family approach is well understood by all parties involved prior to the execution phase and during the execution phase and review/finalized approval phases when validation is involved, while ensuring that validation is not treated like a “paper mill”.
In Part 3 of this article series, we will discuss:
- A technically inexperienced senior global QA requests a protocol deviation instead of accepting the comment “as is”.
- Inconsistent documentation upon executing the “Utilities Verifications” section/attachment of the protocol.
- The execution results stipulated in the output (Actual Result column) do not consistently match the corresponding evidence record.
References
About The Authors:
 Allan Marinelli is the president of Quality Validation 360 Incorporated and has more than 25 years of experience within the pharmaceutical, medical device (Class 3), vaccine, and food/beverage industries. His cGMP experience has cultivated expertise from manufacturing/laboratory computerized systems validations, computer software assurance, information technology validation, quality assurance, engineering/operational systems validation, compliance, remediation, and other GxP validation roles controlled under FDA, EMA, and international regulations. His experience includes commissioning qualification validation (CQV); CAPA; change control; QA deviation; equipment, process, cleaning, and computer systems for both manufacturing and laboratory validations; artificial intelligence (AI)-driven embedded software and machine learning (ML); quality management systems; quality assurance management; project management; and strategies using the ASTM-E2500, GAMP 5 Edition 2, and ICH Q9 approaches. Marinelli has contributed to ISPE baseline GAMP and engineering manuals by providing comments/suggestions prior to ISPE formal publications.
Allan Marinelli is the president of Quality Validation 360 Incorporated and has more than 25 years of experience within the pharmaceutical, medical device (Class 3), vaccine, and food/beverage industries. His cGMP experience has cultivated expertise from manufacturing/laboratory computerized systems validations, computer software assurance, information technology validation, quality assurance, engineering/operational systems validation, compliance, remediation, and other GxP validation roles controlled under FDA, EMA, and international regulations. His experience includes commissioning qualification validation (CQV); CAPA; change control; QA deviation; equipment, process, cleaning, and computer systems for both manufacturing and laboratory validations; artificial intelligence (AI)-driven embedded software and machine learning (ML); quality management systems; quality assurance management; project management; and strategies using the ASTM-E2500, GAMP 5 Edition 2, and ICH Q9 approaches. Marinelli has contributed to ISPE baseline GAMP and engineering manuals by providing comments/suggestions prior to ISPE formal publications.
Abhijit Menon is a professional leader and manager in senior technology/computerized systems validation while consulting in industries of healthcare pharma/biotech, life sciences, medical devices, and regulated industries in alignment with computer software assurance methodologies for production and quality systems. He has demonstrated his experience in performing all facets of testing and validation including end-to-end integration testing, manual testing, automation testing, GUI testing, web testing, regression testing, user acceptance testing, functional testing, and unit testing as well as designing, drafting, reviewing, and approving change controls, project plans, and all of the accompanied software development lifecycle (SDLC) requirements.