
The Value Of Standardizing Risk Assessment Across Quality Systems
By Moria Feighery-Ross, Pharmatech Associates

In recent years, the trend in the pharmaceutical, biopharma and device industries has been to devote more attention to holistic quality systems and to ensure standardization of the company’s overall quality systems rather than using process-by-process policies that may or may not align completely. The 2015 changes to ISO 9001 and ISO 14644-1 and -2 standards have magnified the industry’s focus on risk assessment and quality management systems, integrating and unifying our approach to ensuring quality across all aspects of pharmaceutical, biopharma, and medical device manufacturing.
We are all familiar with risk assessment, which frequently takes the form of failure modes and effects analysis (FMEA) for process and/or equipment; we assess personnel risk with hazard and operability studies like HAZOP; we assess risk in device history and when we choose which instruments in a system to calibrate or which alarm conditions in the building management system should send out alerts. We have always used risk as a selection criterion in the context of environmental monitoring, alarm criticality, and supply chain management, but we didn’t always document it as such. Only recently has our industry begun to codify all these criteria into formalized risk assessments in procedure and practice.
Background Of Revisions
For the purposes of this discussion, the ISO 9001 revision can be summed up in two related topics, the first of which is an increased emphasis on risk-based thinking in general. The second is a reduction in specific prescriptions for quality systems, coupled with the addition of requirements that broaden the scope of documented decision making into all aspects of the business. In short, these changes are less specific about what we must do, but show that we must be able to defend when and why we made those decisions. While it’s true that most U.S. companies in the pharma and biopharma industries do not audit for compliance to ISO 9001, it is an international standard that we lean on for guidance and best practices.
While the ISO 14644-1 and 14644-2 revisions are more specific to environmental air sampling and monitoring in controlled environments, we can apply their lessons to other areas just as we can apply risk assessment to other factors of operation. In 14644-1, the revisions have simplified determining the number of sampling locations and are less prescriptive with respect to locations of same; they have removed the requirement for 5.0µ particle monitoring as well, but we still need it as an early indicator of microbial contaminants and for European market products. In ISO 14644-2, the updates emphasize the need for an ongoing monitoring strategy using risk assessment based on the space’s utilization to better assess and improve clean area operation over time.
Codifying Risk Assessments — Examples
I’ll present some examples from my recent experience and that of my colleagues on the topics of viable and nonviable particulate monitoring, supply vendor assessments, process design, and CMO selection. I invite more discussion and examples in the comments.
Particulate Monitoring
In the past, while selecting sample sites for environmental particulate monitoring, we’ve kept risk at the forefront of our minds as part of the decision process. We know that sites are riskier when they have more personnel contact and that other risk factors such as airflow patterns come into play. But we also need to be able to defend our selection of sample sites, and that part is critical.
When we sat down to codify our consideration of the various risks for viable particulate contamination, we determined the three most important risk factors as: proximity of the location to critical materials and/or tasks; the materials’/tasks’ degree of exposure to the local environment of the location; and the dwell time of the process in the location. In short: how close, how much, and how long? Using these risk factors to generate a matrix with standardized evaluation criteria, not unlike a process FMEA, sometimes led us to different choices than we would have made with the ad hoc determinations.
In the example below, based upon a biotherapeutics process, two non-numerical tables are used to assess risk as an aid to determining placement of EM sample sites. The first table, Table 1A, categorizes the product’s degree of exposure to the environment. The second, Table 1B, is a framework for assessment of different locations within the controlled area under consideration, according to the types of activities performed within. Each organization should make its own unique determination of how the results of these two categories are weighted and combined with other existing specifications to achieve a final answer.
Table 1A: Product Exposure

Table 1B: Activity and Proximity
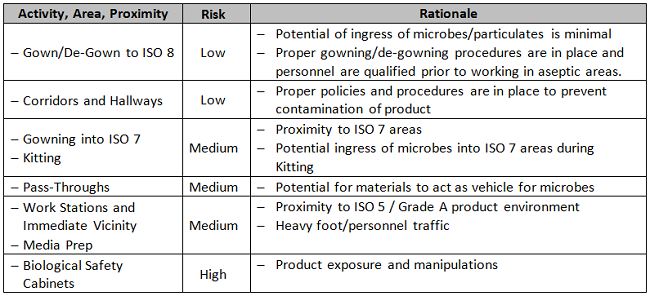
Supply/Services Vendor Assessment
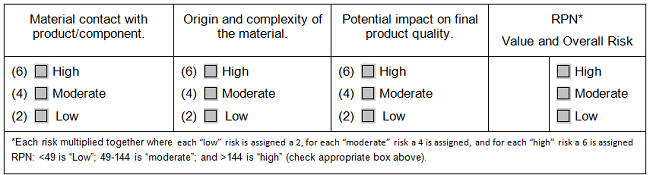
In supply chain management, just as in environmental monitoring, companies use the idea of risk to determine the intensity of supplier scrutiny, but this consideration was largely thought of as the importance of the supplier’s goods or services to the final product. As before, when we sat down to codify it, we gained clarity and focus during the selection process. How complex are the goods or the service involved — such as purified water vs. cell culture media, calculators vs. control systems? What is the origin of the goods/service — such as single-source vs. multi-source, local vs. overseas? And finally, what potential exists for direct quality impact on the final product — if the goods or services are of uncertain quality, how strongly could that affect our final product?
An assessment like this does not have to look like a FMEA. It can be a heat map with simplified categories or (see Table 2) it can simply be a series of columns with predefined meanings for what low, medium, and high risk are within each of the categories. The categories can be used for decisions on what level of scrutiny is appropriate for a potential vendor. This table does not discuss how to use the results of the assessment: Each company, each quality system, each process will have different assignments of low, medium, or high risk based upon the company’s own experience of where goods or services may fall short and what is critical for the success of the process.
Table 2: Selection Matrix for Materials Vendor Assessment
Process Design
Increasing agency scrutiny has also encouraged risk assessment as part of process design, because it becomes part of design history documentation. Within process design, individual risk assessments may feature in equipment and materials selection, parameters and specifications, unit operation development, tech transfer, design of workspaces and/or facility upgrades, and generation of procedures for use in the process.
Figure 1 shows a basic assessment of risk to a list of critical quality attributes (CQAs) for each of five unit operations for an oral solid dosage product.1
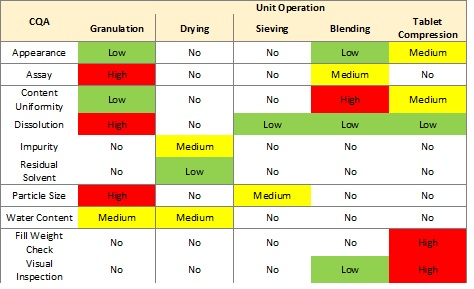
Figure 1: Unit operation risk table
Contract Manufacturing Organization Selection
As with many other business processes in this industry, we have used risk assessment as part of the selection process for a CMO. But again, as with other processes, these assessments have not always been formally documented. The team can classify a CMO along a series of parameters, and then assign a weight to each factor to arrive at the best solution with the available information. This assessment could look quite similar to the vendor selection table in Table 2, but would clearly have many more requirements and considerations, such as:
- Manufacturing ability, familiarity, and equipment for a specific type of product
- Compliance history with agencies and governing bodies
- Quality management systems adequacy
- Manufacturing capacity
- Regulatory history
- Business longevity
- Location
- Laboratory and analysis availability
Resolution
Development, validation, regulatory approval, compliance, and continuous improvement are all features of the overall process of manufacturing and releasing product that can be enhanced by proper use of risk assessment. The additional documentation that the regulatory agencies’ increasing focus on risk assessment has encouraged does raise the complexity hurdle, especially for new companies with few employees and evolving infrastructure. Even for established companies, there are challenges, such as how to not lose the effort and information developed by formal risk assessment within the forest of other documentation that sprouts around each product and within each department. All sizes of companies must address these (and more) added considerations: Where do you keep that documentation and who is responsible for maintaining and controlling it? How can we continue to use this information moving forward?
Even with the additional effort required and increased complexity, increasing focus on risk assessment and risk management and fostering a company culture of continually considering risk can only improve our overall product quality and sharpen our ability to serve our patients and other stakeholders at the highest level.
References:
- Figure 1 is adapted from “Process Design & Risk Management — A Proactive Approach,” by S. Wassink, 2017, Pharmaceutical Online. https://www.pharmaceuticalonline.com/doc/process-design-risk-management-a-proactive-approach-0001
About The Author:
 Moria Feighery-Ross, CQA, is a validation project manager with Pharmatech Associates. She has worked in the regulated life sciences industry for over 10 years. With her technical expertise in validation and quality systems, she provides Pharmatech’s clients with end-to-end support in new facility startup and validation. Feighery-Ross holds a B,S in biology from the University of California, San Diego.
Moria Feighery-Ross, CQA, is a validation project manager with Pharmatech Associates. She has worked in the regulated life sciences industry for over 10 years. With her technical expertise in validation and quality systems, she provides Pharmatech’s clients with end-to-end support in new facility startup and validation. Feighery-Ross holds a B,S in biology from the University of California, San Diego.