
Process Analytical Technology: Meeting FDA's Aspirational Guidance In Continuous Manufacturing
By John D. Orr, Ph.D. and George L. Reid, III, Ph.D.
Ensuring medicinal product quality is our industry’s responsibility, and the FDA has written that “quality cannot be tested into products; it should be built in by design.” In 2004, the U.S. FDA published an aspirational guidance for industry on Process Analytical Technology (PAT) for the routine use of PAT in an integrated manner to design, measure, and control critical process quality and performance attributes through timely and appropriate measurements. The use of well-designed and robust processes and analytical testing strategies, including PAT, enables the industry to confidently ensure product quality.
The first article of this three-part series provided an introduction to PAT and discussed the challenges associated with its adoption and implementation by industry. One challenge noted was the lack of substantial corporate exposure to PAT. The second article introduced the types of analytical tools that are used in PAT and offered approaches for the industry to gain experience and implement in low-risk applications. Examples illustrated where traditional analytical tests could be supplemented, improved, or replaced with PAT tools, such as identity testing, concentration verification, and concentration monitoring (in real time). In these examples, the emphasis was placed on the “AT” in “PAT.”
This article will describe drug substance and drug product examples where industry has succeeded in meeting the FDA’s guidance. (Please note that this overview of continuous manufacturing DS and DP processes enabled by PAT does not discuss the significant development and regulatory activities required to obtain the marketing approval from regulatory authorities.)
PAT For Process Control
In the 14 years since the agency’s guidance was published, the adoption and routine use of PAT in the spirit of the guidance has only slowly been realized. In most cases, utilization of traditional manufacturing (step-by-step, batch processes) and analytical controls in existing factories, with existing equipment, resulted in the production of material of acceptable quality and yield. With batch processing, typically one process step (e.g., chemical reaction, drug product unit operation) is performed and resultant materials are isolated. Subsequent steps are performed in this fashion, until the desired material (drug substance or drug product) is obtained. These manufacturing recipes have process set point conditions or ranges, and they use in-process and final testing to confirm (and control) material quality. This is referred to as quality by testing. Pharmaceutical development using a quality by design (QbD) approach (International Conference on Harmonization [ICH] guidance documents Q8 – Q11) has led to improved process understanding (e.g., material properties, process parameter impact on downstream product attributes) and quality.
Ready to integrate Quality by Design into your development process? Learn the differences between the old GMP mindset and the new quality systems mindset – and how they both affect our daily operations in the course:
Quality by Design (QbD): Making Sense of the ICH Q8, Q9, Q10 Puzzle
PAT integrates analytical tools, informatics, and data modeling to aid process understanding. Pharma companies, in collaboration with vendors, have succeeded in developing robust PAT toolboxes to probe processes. Adding the ability to analyze and adjust a process in real time using PAT allows product quality to be continuously assured. In practice, however, the need for PAT in a commercial setting is minimized by a thorough knowledge of the process, technology transfer to the plant, validated processes, and analytical controls.
One part of the FDA’s mission is to promote public health by reducing drug shortages caused by quality and manufacturing problems. A guidance for the advancement of emerging technology applications for pharmaceutical innovation and modernization was published in 2017. The FDA notes that emerging technology is critical to improving quality and notes that this may represent challenges to both the industry and the agency.
One emerging manufacturing technology that has spurred the use of PAT within the pharma industry is continuous manufacturing (CM). CM has significant allure to pharma and support from the FDA.1,2,3,4 There are many potential benefits to a CM process, including reduced equipment scale and plant footprint, faster scale-up and technology transfer, and a reduction in the amount of materials used and waste generated during process and formulation development. This leads to a lower cost to develop a product and to build and maintain a commercial facility. Pharma and manufacturing equipment vendors are collaborating on the development of portable, continuous manufacturing pods. These are quick to set up and can be used to develop the product. Subsequently, once the product development is complete, the same continuous manufacturing equipment can be transported to the site where the commercial supplies will be manufactured. The technology is transferred by transferring the actual equipment in the pod. The “scale-up” in this case is accomplished by simply running the process for a longer time. Few, if any, problems that are typically encountered with batch process transfer and scale-up will be encountered with a CM process.
In CM, materials are metered into the process regularly and the product is generated repeatedly. There is typically a small residence time for the ingoing materials to be transposed into the desired product (e.g., drug substance, intermediate, drug product blend, finished dosage form). As the desired material is produced nonstop, there is a potential to continuously verify the material quality and adjust the process parameters to maintain a consistent product quality. PAT provides the ability to add analytical technology to monitor a process in situ, enabling CM with real-time measurements of the process. To embrace the FDA’s PAT guidance, pharmaceutical companies needed a key initiative and business driver. The technological advances in manufacturing and analytical technology have made continuous cGMP drug substance and drug product manufacturing feasible. In the following examples, the application of PAT to aid CM will be discussed. The first example describes how PAT is utilized in support of the CM of a cGMP small molecule drug substance. The second example discusses the PAT control systems in the manufacture and real-time release of a commercial drug product.
Drug Substance Continuous Processing
A multistep continuous-flow cGMP process was set up in laboratory fume hoods, and it produced a small molecule drug substance [prexasertib monolactate monohydrate (12; Figure 1)] suitable for use in human clinical trials.5,6 Eight unit operations performed in small continuous reactors, extractors, evaporators, crystallizers, and filters were integrated and resulted in the manufacture of 24 kg of drug substance at a rate of 3 kg/day.
The process utilized to produce 12, in four steps, from an advanced intermediate (7) is shown in Figure 1. Step 1 is a reaction of a Boc-protected compound with a highly toxic and reactive reagent, hydrazine, in a flow-through reactor resulting in pyrazole formation (8). Step 2 is a nucleophilic aromatic substitution reaction with a chloropyrazine (9). Step 3 removes the Boc protecting group in an acid-catalyzed reaction. Finally, Step 4 (when the lactate salt is desired) is the salt formation reaction.
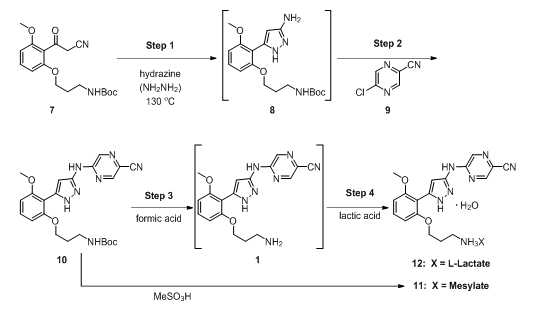
Figure 1: Chemical route used to make 12 in continuous flow.
Performing the chemistry in a continuous flow mode (illustrated in Figure 2) allowed key issues associated with batch mode to be overcome. For example, Step 1 was the condensation of 7 with hydrazine. In batch mode, this reaction required a large excess of hydrazine and extended reaction times. This posed serious risks, as hydrazine is highly toxic and explosive.7,8 In fact, the mesylate salt of prexasertib (11) was previously manufactured by a nine-step route that was deemed unsuitable for long-term manufacturing due to the use of several hazardous reagents.5 Conducting the reaction in a small-volume stainless-steel pressure reactor, with only a slight excess of hydrazine at an elevated temperature, afforded rapid production of highly pure product (8) in high yield.9 Hydrazine was safely used since only a small portion of it was present at any time in the reactor. An additional benefit was that residual unreacted starting material (SM, 7) levels in product (8) were typically in the range of 0.05 — 0.20 percent — because of the high reaction efficiency resulting from continuous manufacturing flow use.
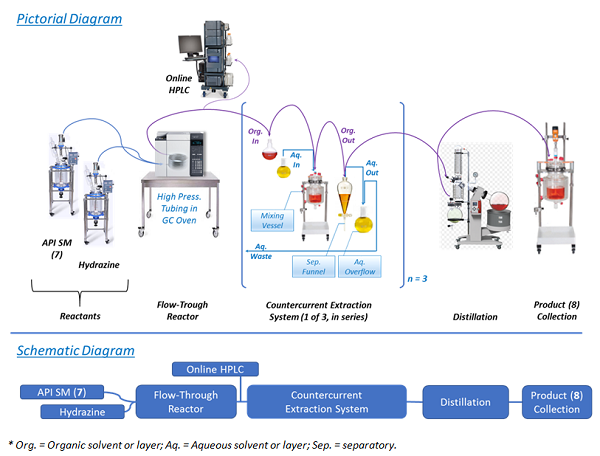
Figure 2: Pictorial and schematic diagrams of process Step 1
Following the Step 1 reaction, purification of the product (8) and exchanging of the solvent were performed. This was accomplished using continuous countercurrent extraction as diagrammatically illustrated in Figure 2 and explained, based on chemical principles, in Figure 3.
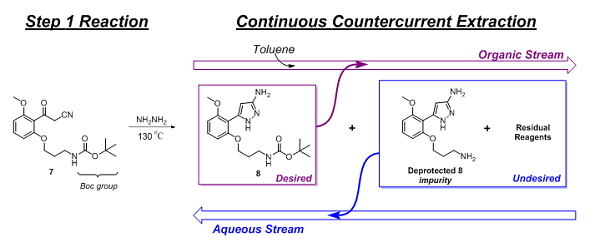
Figure 3: Purification and solvent exchange of Step 1 product via continuous countercurrent extraction
The main reaction impurity, deprotected 8 and residual reagents (hydrazine and acetic acid) were extracted and removed by the aqueous stream, while 8 flowed forward in the organic stream. Initial levels of the deprotected 8 impurity, following the reaction, were as high as 5 percent but were reduced to approximately 0.5 percent by this continuous purification step. Additionally, residual hydrazine levels were reduced to less than 2 ppm. Finally, chromatographic analysis showed 8 to be 98.25 percent (HPLC area-%) pure.
The use of PAT was crucial to the development and execution of this continuous process. It was used extensively during development for process characterization. During cGMP production, PAT provided data that was used to ensure and demonstrate that the process operated in a state of control, as designed. Continuous processing allowed the manufacturer of high-purity cGMP API, at the rate of 3 kg/day, in a safe and cost-effective fashion within laboratory fume hoods. The use of online PAT was a key component enabling this breakthrough accomplishment.
Similar advances in the continuous manufacturing of cGMP drug product have been enabled using PAT, as well, as described in the example below.
Drug Product Continuous Processing
The manufacture of solid oral drug products via continuous processes represents a compelling opportunity. The challenges posed by adoption of continuous DP processing are somewhat reduced compared to continuous drug substance processing. For example, there are a limited number of standard major unit operations (dispensing, blending, granulation, drying, milling compression, coating, and packaging), and process parameters are similar among different drug products. Further, some operations are already operated in continuous mode (e.g., tableting). Costs associated with batch mode DP processing are strong motivators for continuous DP processing adoption. For example, process development, process understanding, scale-up, and technology transfer are time, resource, and material intensive. The potential to better utilize staff and minimize the consumption of raw materials (including the drug substance), while delivering a quality medicine is enticing, and pharmaceutical companies and regulatory agencies are embracing the approach.
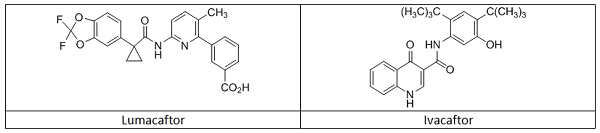
In this example, Vertex’s Orkambi is highlighted as a CM drug product produced utilizing a PAT control strategy. Orkambi contains two active pharmaceutical ingredients, lumacaftor and ivacaftor (Table 1), in a fixed dose combination tablet.10 The medicine is approved in multiple global markets for the treatment of cystic fibrosis, and the CM and use of PAT for control and (real time) release of the product have been thoroughly inspected and reviewed.
Table 1: Structures of the Two Active Pharmaceutical Ingredients in Orkambi
During continuous manufacturing of the drug product, the excipients are continuously dispensed, blended, wet granulated, dried, milled, extra-granular blended, compressed, film coated, and printed. Three sites are reported to produce the drug product using a continuous wet granulation process, and the systems and PAT capabilities differ, only slightly, from site to site.11
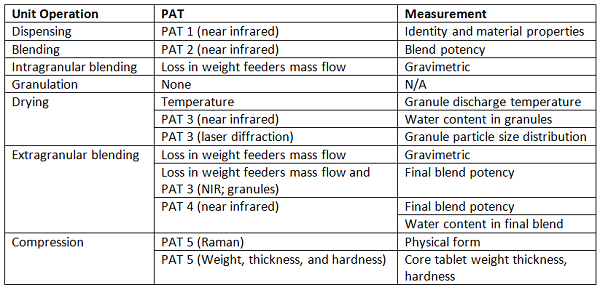
Shown in Figure 4 is a system that operates in continuous mode from individual component feeding in to film-coated tablets, at one of the production sites,12 and shown in Table 2 is an associated list of the unit operations, the PAT tools, and the measurements made. The individual measurements are combined in a control strategy that allows the entire process to be continuously monitored. This enables process adjustments to be made, as needed, to ensure material conformance to specifications, and enables segregation of nonconforming materials.
Design of experiments was performed to understand the impact of unit operation parameters and material attributes with subsequent operations. This linking of the individual unit operation process parameters and material attributes allowed for feed-forward and feedback control, ensuring that the final product quality requirements were met.

Figure 4: Vertex-GEA CM rig.13 1. At-line NIR incoming material attributes; 2. NIR blend potency; 3. Granule properties (3a. NIR granule uniformity, moisture; 3b. Laser diffraction particle size); 4. NIR final blend, potency and moisture; 5. Tablet properties (5a. Raman API form and identification; 5b. Weight, thickness, hardness); 6. Raman film coat thickness.
Table 2: PAT Application by Manufacturing Unit Operation
 /p>
/p>
Conclusions
The uptake and adoption of PAT post the FDA’s 2004 guidance were slow due to a number of factors discussed in the first article. Pockets of scientists did learn and apply these analytical tools, leading to improved knowledge and process understanding, as described in the second article. These early adopters also purchased equipment and worked with vendors to improve the technology to its current state.
Continuous manufacturing is an ideal application to marry the process and analytics. Real-time process measurements are necessary to confirm and control product quality, and it has led to an increased adoption of PAT within the industry. Publications and themed conferences are leading to more informed and educated scientists and corporate stakeholders. Continuous manufacturing is a current and future state for industry, and PAT will continue to enable this initiative. Industry has attained the FDA’s aspiration guidance as described in 2004.
References:
- D. am Ende. Process Design toward Continuous Manufacturing: a Green Perspective. 2009 Green Chemistry & Engineering Conference.
- S. Chatterjee. FDA Perspective on Continuous Manufacturing. 2012 IFPAC Annual Meeting.
- S. Neil. Pharma Catches on to Continuous Manufacturing. Automation World. December 6, 2017.
- A.E. Hagrasy. Scientific and Regulatory Considerations for CM Implementation for Drug Product. 2017 PQRI.
- Cole, K. P; Groh, J. M.; Johnson, M. D.; Burcham, C. L.; Campbell, B. M.; Diseroad, W. D.; Heller, M. R.; Howell, J. R.; Kallman, N. J.; Koenig, T. M.; May, S. A.; Miller, R. D., Mitchell, D.; Myers, D. P.; Myers, S. S.; Phillips, J. L.; Polster, C. S.; White, T. D.; Cashman, J.; Hurley, D.; Moylan, R.; Sheehan, P.; Spencer, R. D.; Desmond, K.; Desmond, P., and Gowran, O. Science. 2017, 356, 1144-1150. DOI: 10.1126/science.aan0745.
- As the process chemistry was being developed for 11, the mesylate salt (12) was found to have improved aqueous solubility and was identified as the desired commercial salt form.
- http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM464285.pdf
- http://www.cdc.gov/niosh/ipcsneng/neng0281.html
- Flow-mode optimization studies showed a mean reaction residence time of 60 min in 500 psi gauge tubing at 130 °C was ideal. This was conducted with the use of use of a gas chromatography (GC) oven.
- Orkambi prescribing information.
- Orkambi European public assessment report.
- Orkambi FDA chemistry reviews.
- H. Thomas “Control Strategy Implementation Considerations for a Continuous Drug Product Manufacturing Process,” Product Quality Research Institute, October 05, 2015.
About the Authors:
 John D. Orr, Ph.D., president of Associated Analytics LLC, has 25 years of pharmaceutical chemistry, manufacturing, and controls (CMC) experience. He founded Associated Analytics in 2017, serving clients in need of analytical chemistry expertise. Orr was previously with Rondaxe, where he assisted clients in the areas of CMC analytical and process development, and Eisai, where he served as director of analytical and physical chemistry and as a member of the team that developed and commercialized complex natural product-based medicines.
John D. Orr, Ph.D., president of Associated Analytics LLC, has 25 years of pharmaceutical chemistry, manufacturing, and controls (CMC) experience. He founded Associated Analytics in 2017, serving clients in need of analytical chemistry expertise. Orr was previously with Rondaxe, where he assisted clients in the areas of CMC analytical and process development, and Eisai, where he served as director of analytical and physical chemistry and as a member of the team that developed and commercialized complex natural product-based medicines.
Orr has been active in several industry groups, serving on the board of directors of the IQ Consortium and the Allotrope Foundation. He received his chemistry degrees from the University of Pittsburgh (B.S.) and The University of Chicago (M.S. and Ph.D.), and performed post-doctoral studies in molecular biology at The Noble Foundation. You can reach him at john.orr@AssociatedAnalyticsLLC.com or connect with him via LinkedIn.
 George L. Reid, III, Ph.D., principal consultant with Cardinal Health Regulatory Sciences, has more than 20 years of pharmaceutical CMC technical and regulatory experience, supporting projects from preclinical to post-approval. He was previously with Pfizer, in project and technical support roles (PAT, separations science, and technology development). He has contributed to multiple approved medicines and currently works with clients in the areas of analytical chemistry, drug substance and formulation development, and compliance audits.
George L. Reid, III, Ph.D., principal consultant with Cardinal Health Regulatory Sciences, has more than 20 years of pharmaceutical CMC technical and regulatory experience, supporting projects from preclinical to post-approval. He was previously with Pfizer, in project and technical support roles (PAT, separations science, and technology development). He has contributed to multiple approved medicines and currently works with clients in the areas of analytical chemistry, drug substance and formulation development, and compliance audits.
Reid has been active in industry groups, including the IQ Consortium analytical leadership group and PAT lead, and currently is on the planning committee for the Land O'Lakes Pharmaceutical Analysis Conference. He earned a doctorate in chemistry from Missouri University of Science and Technology and a B.S. in biochemistry from Beloit College. You can reach him at george.reid@cardinalhealth.com or connect with him via LinkedIn.