
When Does GMP Matter In Non-GMP Settings?
By Takehiro Okumura and Zeb Khan, Takeda Pharmaceuticals

Regulatory agencies allow for manufacturing materials such as genome editing materials, the generation of iPS cells, and the subsequent selection process under principles of GMP or GMP-like operation instead of full GMP compliance.1,2,3,4 In Japan, the Pharmaceuticals and Medical Devices Agency (PMDA) does not specify whether to use GMP or non-GMP during the clinical phase and the clinical study sponsor owns the responsibility to secure the product quality for patients.
This may sound interesting for biotech ventures or scientists who are working in discovery organizations. It means that you can conduct upstream manufacturing of materials or intermediates in your labs once you developed the process for them without performing technology transfer to an internal or external GMP-compliant organization. The tech transfer process may consume valuable time and oftentimes may not be successful due the complexity and uncertainties of manufacturing. If scientists who developed the manufacturing processes could perform the manufacturing by themselves in their labs, it could save significant time and resources and increase the likelihood of success in manufacturing clinical supplies.
This article explains how to implement GMP principles in non-GMP settings, also known as GMP-like, and the quality oversight required for implementation.
Implementing Principles Of GMP In An R&D Lab
First, it is significant to understand the rationale for manufacturing or processing material in a non-GMP workspace while following the principles of GMP. This shall be assessed based on an understanding of how the material will be used and the regulatory strategy governing the overall program. One can ask the project owner the following questions:
- What is the project status? Is it preclinical, clinical, or pre-candidate selection, etc.?
- Why is manufacturing/processing in a GMP environment not feasible?
- Is the request benefiting cost, timelines, efficiency gains, or patient urgency?
- What is the impact on short-term vs. long-term goals for the project?
- What is the regulatory strategy supporting the request?
- Has the value proposition for manufacturing using principles of GMP been assessed in terms of risks?
- Does the risk assessment include cross-functional collaboration?
- Are the manufacturing process steps and relationship to the final product manufacturing understood?
Applicability of GMP or “principles of GMP” should be carefully assessed by quality and CMC regulatory experts based on the interpretation of guidance from regulatory agencies.
The application of quality risk management (QRM) principles is crucial in determining the necessary level of GMP for the manufacturing of a product that is intended to be used as clinical trial material. The European Medicines Agency (EMA) has provided guidance on the matter stating that manufacturers should adopt a risk-based approach to identify the relevant GMP requirements necessary to ensure the quality, safety, and efficacy of the final drug product while considering the associated risks. It is, therefore, incumbent upon manufacturers to apply this approach to adequately mitigate any potential risks to the quality, safety, and efficacy of the final product.
It would be acceptable to use an R&D lab with appropriate modifications instead of using a GMP-compliant ISO-certified sterile production facility as long as all the risks are duly understood and measures are put in place to eliminate the risks or mitigate them to an acceptable level.
For example, if you are confident that you can detect microbial contamination during long-term cell culture and/or downstream steps with robust quality control for the final drug product, the following mitigations would be sufficient to reduce the risk for microbial contamination during manufacturing in the R&D lab:
- attach a HEPA filter for air supply,
- set differential room pressure against adjacent rooms,
- dedicate the use of reagents and ancillary materials, and
- establish a gowning procedure, etc.
Aseptic process simulation tests with the most junior operators may help to evaluate the risks of failure of production.
Regarding risk assessments for cross-contamination, microbial/virus contamination, or operational failure, a cross-functional team should be formed including manufacturing, analytical, and quality. Microbiologists would help to ensure a thorough assessment. Based on the risk assessment and overall evaluation, the final decision to utilize a non-GMP workspace under the requirements of the principles of GMP must be assessed in collaboration with regulatory experts.
Agreed mitigations such as facility modifications and the establishment of a relevant quality management system are written in a quality project plan that defines concrete control levels/procedures and mitigations for individual quality system elements. Quality oversight must be included for each control.
A cross-functional team including quality shall confirm on-site that all risks are addressed per the QRM mitigation plan before starting operations at the facility/lab. The overall approach is outlined in Figure 1.
Baseline requirements for the principles of GMP are outlined in Appendix 15,6 as an example. Gaps in the requirements for GMP principles should be evaluated and mitigated appropriately.
Figure 1. Process flow of implementation of “principles of GMP” in non-GMP workspaces
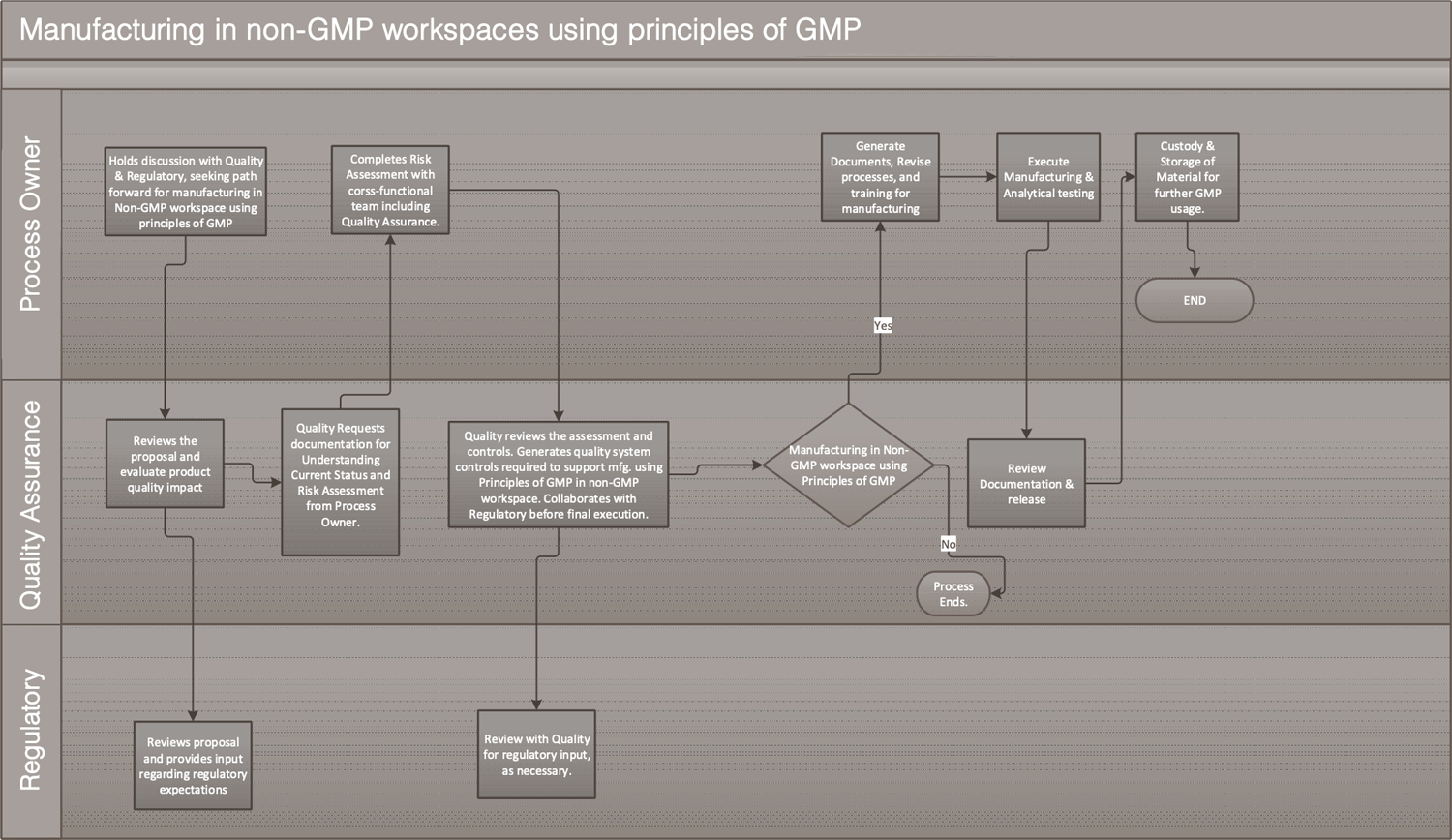
Click for full-screen view
General controls:
- Establish written procedures/work instructions for manufacturing, including personnel training, equipment maintenance, and testing as applicable.
- Ensure employees are trained in GMP principles and aware of their responsibilities in maintaining a GMP-compliant manufacturing environment.
- Establish control over the quality of materials and storage.
- Maintain accurate and complete records of all aspects of the manufacturing process and any incidents or nonconformities.
- Ensure that the manufacturing facility, equipment, and utensils are maintained to minimize the risk of contamination.
It will require intensive resources, hands-on support, and guidance by quality professionals rather than usual operational quality support under the established GMP quality system.
Further Risk Mitigations
When starting renovation activities to the controlled lab, the manufacturing process and control may not be fully finalized. However, once the actual operational procedures are defined, a thorough risk assessment for individual processes with blank batch records would identify any potholes to be addressed. For example, cross-contamination through common equipment used in adjacent non-controlled labs can be easily mitigated.
Execution Of Operation
The frequency of manufacturing campaigns utilizing the controlled lab may dictate the need for detailed operating procedures. To streamline operations and ensure a traceable record for future regulatory filings, it may be prudent to define specific procedures in a product-specific document, such as the product master formula and its blank batch record. This approach will minimize the number of operating SOPs and provide an executed batch record that captures all actions. Such an approach will also help to reduce operating errors by employees working in the controlled lab.
Quality Control
For the product manufactured in the controlled lab under the principles of GMP, appropriate material specifications consisting of critical attributes should be established ahead of manufacturing and tested for further use by a GMP-compliant testing laboratory using appropriately qualified testing methods.
The challenges to establishing appropriate specifications arise from various factors, and uncertainties in the early development phase are unique to this industry. For example, induced pluripotent stem cell engineering takes a long time (e.g., more than a year) for multiple gene editing steps followed by single-cell cloning, banking, and further cell expansion to get sufficient cell numbers for the final drug product. At the stage of manufacturing of genome editing materials, manufacturing steps using these materials and/or downstream process development of the final product may still be underway. Various types of raw materials like organic chemicals, protein, material of biological origin, research-grade material, and non-sterile material will be used in manufacturing in the controlled lab; however, the impact of these materials’ quality attributes on manufacturability and quality of final drug product is not fully understood in early development.
Overall Control Strategies For Cross-functional Teams
Even the most robust production plan could get upended when business decisions — for example if your company chooses a new vendor for materials — happen without an overall quality control strategy. In such a complex and uncertain environment, the appropriateness of material specifications may evolve with process development and with assumptions of the project strategy. So, to manage the project in such challenges, it is recommended to write an overall quality control strategy for the project that defines quality attributes of starting material, cell banks, critical material, and process parameters based on the understanding of the product. A visualized plan for the overall control strategy for setting specifications would help the cross-functional team align with the agreed strategy and the prerequisites of the plan.
Regardless of whether you practice GMP manufacturing or follow the principles of GMP, the importance of managing risks and uncertainty mentioned above is significant and remains unchanged for patient safety. Risks such as microbial contamination, viral or TSE contamination, lot variability, and residual impurities should be thoroughly assessed with an understanding of material point-of-use and functionality. It is imperative to undertake a thorough analysis of risks that can be effectively managed during the production or downstream phases. The identification of such risks is crucial to mitigate any potential negative impact on the project's outcome. For example, microbial contamination can be detected during long-term incubation of cells, and sterility tests can be carried out using rapid methods at multiple steps during production. Take note: viral contamination is not completely detected in the downstream phase. Therefore, you need to perform a thorough risk assessment for viral safety and follow regional regulatory requirements before any product is used in manufacturing clinical trial material.
Quality Control For Critical Materials
Various materials of GMP/non-GMP grade may be used in the controlled lab. Even if they are used in manufacturing under the principles of GMP, risks for using such raw materials should be assessed as they would in GMP operations since it is the manufacturer’s responsibility to ensure patient safety.
The output of risk assessment for all the raw materials serves as the basis of understanding for the creation of additional measures for use in your controlled lab, for example: sterile filtration, incoming material receipt processes, retention sample, release strategies (e.g., qualification or identity test before use in production, etc.) as well as material specifications. It is recommended to create a written document – a material control plan – as a part of the control strategy.
Figure 2
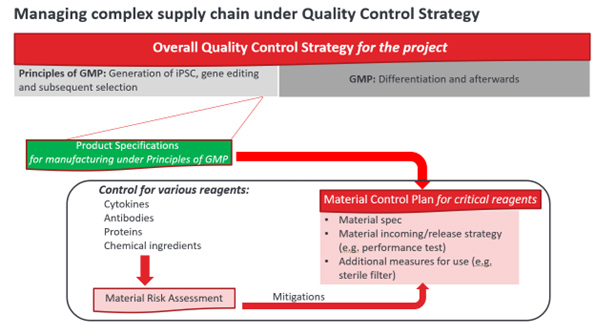
The material control plan should be aligned with the product specifications and overall quality control strategy of manufacturing. When the project strategy is changed during project development, the control strategy must be updated and the associated product specifications and material control plan may be revisited accordingly. In that way, the overall quality control strategy, including downstream operations in GMP manufacturing, will be aligned from end to end and kept updated appropriately — even in endless environmental changes. The governance of overall quality control is illustrated in Figure 2.
The contents of this publication represent the views of the presenter and do not necessarily reflect the views of the author’s employer.
References:
- EMA guidance: Questions and answers on the principles of GMP for the manufacturing of starting materials of biological origin used to transfer genetic material for the manufacturing of ATMP (EMA/246400/2021)
- EMA, Guideline on quality, non-clinical and clinical aspects of medicinal products containing genetically modified cells (EMA/CAT/GTWP/671639/2008 Rev. 1)
- PIC/S Annex 2A (see “Principles of GMP” on page 56)
- FDA CBER OTAT town hall: Gene Therapy Chemistry, Manufacturing, and Controls (September 29, 2022), transcription and recording are available in URL as of 22Jan2024 OTAT Town Hall: Gene Therapy Chemistry, Manufacturing, and Controls - 09/29/2022 | FDA
- FDA Guidance for Industry CGMP for Phase 1 Investigational Drugs (July 2008)
- PDA Technical Report No. 56 Revised 2016 (TR 56) Application of Phase-Appropriate Quality System and cGMP to the Development of Therapeutic Protein Drug Substance (API or Biological Active Substance)
About The Authors:
 Takehiro Okumura, Ph.D., is a director of Cell Therapy Quality at Takeda. He has more than 25 years of experience in the pharmaceutical industry working from R&D to commercialization, including analytical development, regulatory submissions, quality system and compliance, QC/QA, lifecycle product management. Over the past six years, he built capability for cell therapy quality at from scratch Takeda Boston and Japan and equipped them with in-house manufacturing capacity for cGMP in Boston and GMP principles in Japan. He supported multiple novel autologous CAR-T therapies and one allogenic CAR-NK therapy to first-in-human trials.
Takehiro Okumura, Ph.D., is a director of Cell Therapy Quality at Takeda. He has more than 25 years of experience in the pharmaceutical industry working from R&D to commercialization, including analytical development, regulatory submissions, quality system and compliance, QC/QA, lifecycle product management. Over the past six years, he built capability for cell therapy quality at from scratch Takeda Boston and Japan and equipped them with in-house manufacturing capacity for cGMP in Boston and GMP principles in Japan. He supported multiple novel autologous CAR-T therapies and one allogenic CAR-NK therapy to first-in-human trials.
 Zeb Khan is the head of quality manufacturing operations, cell therapy, for Takeda Oncology in Cambridge, Massachusetts. With over 25 years of experience in the CMC domain, she has supported development projects ranging from preclinical to commercialization, where she has held varying degrees of responsibility in product development, analytical laboratory, and quality assurance. She has coached several teams in applying quality-by-design principles, which has led to the delivery of successful results-oriented strategies. She holds a M.Sc. in organic chemistry and a master's degree in regulatory science and health safety.
Zeb Khan is the head of quality manufacturing operations, cell therapy, for Takeda Oncology in Cambridge, Massachusetts. With over 25 years of experience in the CMC domain, she has supported development projects ranging from preclinical to commercialization, where she has held varying degrees of responsibility in product development, analytical laboratory, and quality assurance. She has coached several teams in applying quality-by-design principles, which has led to the delivery of successful results-oriented strategies. She holds a M.Sc. in organic chemistry and a master's degree in regulatory science and health safety.