
What Biotech Sponsors Should Look For In A CDMO For Sterile Fill/Finish
By Sarah Sink, Your Pharma Girl

For biotech sponsors, sterile fill/finish is one of the most critical, and riskiest, steps in the drug product life cycle. Unlike other modalities, sterile injectables carry a zero-defect expectation: one missed particle or compromised vial can delay a clinical program and, more importantly, put patients at risk. Choosing the right CDMO partner isn’t about “capacity at a good price”; it’s about a partner with the right mix of technical expertise, regulatory track record, quality culture, and operational transparency. This article focuses on what to prioritize when selecting a sterile fill/finish CDMO and highlights the often-overlooked details that drive timelines, costs, and risk.
Define Your Sterile Needs Before Engaging A CDMO
Before requesting proposals, align internally so CDMOs are evaluated against the right criteria.
- Primary format: Vials, prefilled syringes (PFS), and cartridges each carry distinct regulatory and operational implications.
- Container closure system: Determine whether you need glass or polymer vials, amber vials for light-sensitive products, specialized syringe components for PFS, cartridges, or blowback stoppers for vials to ensure proper sealing. Compatibility among formulation, closure, and container is critical to avoid particulates or integrity failures.
- Lyophilization needs: Do you need lyo for product stability? If so, does the CDMO have the right lyo capacity and experience with similar molecules?
- Batch size: Early trials may need hundreds of units; later phases and commercialization can require tens of thousands.
- Regulatory readiness: Has the CDMO supported INDs, BLAs, or NDAs for similar sterile products?
Sterile fill/finish is where your molecule takes its final form. Clear requirements up front prevent costly misalignment later.
What To Look For In A Strong Sterile Proposal
A credible sterile proposal goes beyond pricing and dates. Look for:
- facility and equipment readiness (filling lines, barrier systems such as restricted access barrier systems [RABS] or isolators, lyophilizers, cold-chain)
- technical assumptions (yields, fill volumes, in-process controls)
- environmental monitoring (EM) history (particulate and microbial control with trending)
- operator expertise (aseptic training, tenure, turnover)
- process validation plan (media fills, aseptic process validation, container closure integrity testing [CCIT] strategy)
- regulatory inspection track record (recent FDA/EMA outcomes and corrective actions).
The Hidden Drivers Of Batch Size
Many teams assume the clinical vial count equals the manufacturing need. In practice, sterile programs consume substantially more product. Drivers of manufacturing volume include:
- retains for QA and potential future testing
- stability studies across time points and, often, multiple orientations (upright, inverted, horizontal) to assess closure performance as each orientation consumes additional units
- analytical method development to refine and qualify/validate a multitude of testing requirement
- formulation development for compatibility and early scale-up adjustments
- PUPSIT (pre-use post-sterilization integrity testing) when sterile filtration is employed
- QC and release testing well beyond sterility and endotoxin:
- full-panel finished product testing per specification (potency, appearance, pH, osmolality, particulates, etc.)
- CCIT using robust, quantitative methods such as vacuum decay or helium leak approaches now preferred over legacy dye ingress
- microbiological assays including bacterial endotoxin testing, sterility, and bioburden
- “for information only” testing requested by CDMO or sponsor teams.
Because these pulls require multiple units (and sometimes multiple orientations or time points), a 2,500-vial plan can quickly become 5,000 to7,000+ vials. That drives API/excipient demand, suite time, and overall cost. Anticipating this early keeps budgets and timelines realistic.
Lyophilization: A Variable That Changes Everything
If your product requires lyophilization, the cycle time can be a major cost and schedule driver. A relatively simple, stabilizer-supported small molecule might have less than a two-day cycle, while a complex biologic could require five or six+ days per cycle, which directly impacts suite occupancy, equipment availability, and cost. Sponsors should ask about:
- expected cycle time for the proposed formulation
- number/scale of lyophilizers and current utilization
- CDMO experience with cycle development/optimization for similar molecules
- how extended cycles are coordinated with stability pulls and analytical testing to avoid delays.
Key takeaway: Longer lyo cycles not only increase price; they can tie up scarce capacity and ripple through the project timeline.
Case Example 1: A Monoclonal Antibody Requiring Lyophilization
A midsize biotech developing an oncology mAb initially planned a 5,000-vial Phase 2 batch. Early stability data showed insufficient liquid stability, so the product moved to a lyophilized presentation. The total need rose to ~12,000 vials after stability pulls (multiple orientations), QC, and retains were included. The six-day lyo cycle further extended suite time and cost. Because the CDMO surfaced these factors early, the sponsor reset API planning, budget, and timeline before commitment.
Lesson: For mAbs, formulation (liquid vs. lyo) can materially change capacity, cost, and schedule.
Quality And Compliance: Nonnegotiable In Sterile Manufacturing
Regulators scrutinize sterile operations intensely. Evaluate both systems and culture, including:
- inspection history and how findings were closed
- deviation management (microbial excursions, sterility failures)
- media fills (frequency, pass rates, acceptance criteria)
- data integrity (ALCOA+ practices in batch records and QC)
- change control around closures, environmental monitoring, and filling parameters.
A CDMO’s quality culture – how they respond when something goes wrong – is the best predictor of real-world performance.
RABS vs. Isolators: A Practical Decision, Not A Binary One
Barrier technology is often reduced to “isolators are best.” In reality, both RABS and isolators are acceptable when properly designed and operated. FDA’s aseptic processing guidance and EMA’s Annex 1 recognize RABS as compliant when robust procedures and monitoring are in place.
Isolators
- Pros: maximum separation, fewer interventions, strong contamination control
- Cons: higher capital cost, longer implementation, complex changeovers
RABS
- Pros: lower cost, easier retrofit, faster to bring online; strong separation with strict SOPs
- Cons: greater reliance on operator discipline; more interventions and monitoring versus isolators
Don’t dismiss RABS. Evaluate performance data (EM trends, media fills), operator training, and inspection history. A well-run RABS line may be as suitable, and more available, than an isolator-only site.
Case Example 2: Small Molecule Sterile Vial Program Using RABS
A small biotech targeting 3,000 vials for a Phase 1 study faced long lead times at isolator-only sites. A RABS facility could start in four months and had strong EM/inspection performance. After a data-driven review, the sponsor chose RABS and launched on time. In retrospect, training, SOP adherence, and transparency mattered more than barrier type, and access to earlier capacity de-risked the trial.
Industry Trends Shaping Sterile CDMO Selection
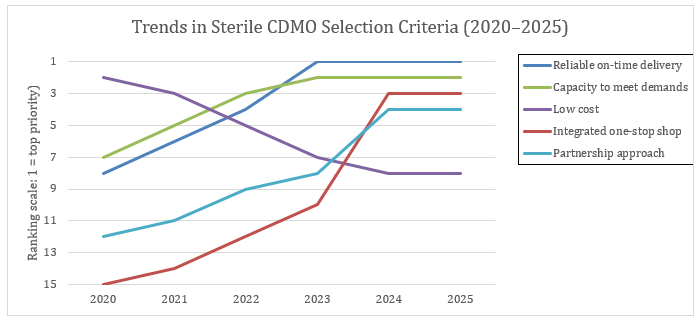
Figure 1: The data shows a clear evolution in how sponsors prioritize sterile CDMO partners between 2020 and 2025. In the early years, cost and established quality records carried the most weight, while on-time delivery and capacity availability were secondary considerations.¹,² The disruptions of the pandemic shifted that balance: by 2023, reliable on-time delivery and capacity to meet demand had become the top two decision drivers, while cost slipped out of the top tier.¹,³ By 2024 and into 2025, priorities expanded further, with sponsors highlighting integrated “one-stop shop” service models and partnership-oriented approaches as defining differentiators.⁴ The result is a new standard for CDMO selection, where the strongest partners are measured not just by technical capability, but by reliability, scalability, and their ability to operate as true extensions of the sponsor’s team.¹,⁴
In 2024, the emphasis continued to evolve, with sponsors also highlighting integrated 'one-stop shop' models and stronger partnership approaches as key differentiators.
Capacity to meet demand rose from 7th to 2nd over the same period.¹ Cost competitiveness fell from #2 in 2020 to outside the top five by 2023.¹,³ By 2024, integrated “one-stop shop” offerings and stronger partnership approaches emerged as critical differentiators.⁴
Net-net: Selection is shifting away from shiny equipment lists toward reliability, scalability, and integration.
Be careful of full one-stop shop integration, though, and remember the old saying of “putting all your eggs in one basket,” as this can cause rippling effects should a bottleneck or disaster occur.
Building A Sterile Evaluation Framework
Keep decisions objective and phase-appropriate:
- Use a weighted scoring matrix across technical fit, quality, capacity, and culture.
- Consider cross-functional participation (CMC, QA, regulatory).
- Perform site visits focused on barrier systems, EM practices, lyo assets, and operator behaviors.
- Review references from similar sterile programs (format, scale, phase).
This approach aligns the CDMO choice with program strategy, not just price pressure.
Partnering For Sterile Success
Sterile fill/finish is the last critical step before your product reaches patients, and it is also the point of greatest regulatory scrutiny. The stakes are too high to treat CDMO selection as a transactional choice. A successful partnership requires a CDMO that combines aseptic discipline, capacity reliability, technical strength, and a culture of quality that can withstand the inevitable challenges of development.
Sponsors that take the time to clarify their internal needs early are better positioned to recognize hidden drivers of batch size, ask informed questions about lyophilization and container-closure integrity, and evaluate barrier technologies on evidence rather than assumptions. These steps not only protect budgets and timelines but also prevent the type of surprises that can derail clinical programs.
Equally important, the right CDMO acts as a strategic extension of the sponsor’s team, not just a service provider. The most effective partnerships are built on transparency, aligned expectations, and open communication. When issues arise, and they always do in sterile manufacturing, a partner with a strong quality culture and responsive communication framework can mean the difference between a manageable deviation and a program delay.
Finally, while capacity, cost, and timelines are important, the ultimate goal is patient impact. Every vial represents a potential dose for someone waiting on a therapy. A CDMO that shares that perspective will prioritize integrity and accountability alongside efficiency.
The bottom line: By choosing a CDMO that emphasizes collaboration, reliability, and quality, biotech sponsors can de-risk their sterile programs, accelerate development, and, most importantly, bring lifesaving therapies to patients faster and with confidence.
References
- Holloway J. Tracking the Evolution of CDMO Selection Criteria for Biologic Drug Substance Manufacturing. ISR Reports. May 8, 2025.
- Allison B. Biopharma Outsourcing Trends for Sterile Injectable Manufacturing. ISR Reports. May 8, 2025.
- Stanton D. CDMO selection and capacity top concerns among biopharma CFOs. BioProcess Insider. February 20, 2023.
- PharmaOffer. Top 10 Biggest CDMO Companies and Market Trends (Key Trends & Insights for 2024). PharmaOffer Blog. 2024.
About The Author:
 Sarah Sink, MBA, is the founder of Your Pharma Girl and an experienced business development leader in the biotech and pharmaceutical services industry. With a background spanning biologics, sterile fill-finish, and oral solid dose development and manufacturing, she has guided numerous biotech sponsors through the complexities of CDMO selection, negotiation, and partnership management.
Sarah Sink, MBA, is the founder of Your Pharma Girl and an experienced business development leader in the biotech and pharmaceutical services industry. With a background spanning biologics, sterile fill-finish, and oral solid dose development and manufacturing, she has guided numerous biotech sponsors through the complexities of CDMO selection, negotiation, and partnership management.