
Understanding CQAs For mRNA/Lipid Nanoparticle Product Development And Manufacture
A conversation with Elaine Peters and Philipp Hoch, BioPhorum

There is a long history of research and development into RNA-based therapeutics and vaccines due to their potential to treat or prevent disease by influencing protein expression. Development activity is now rapidly expanding in the field of mRNA molecules that can drive the expression of therapeutic proteins or antigens, spurred by recent advances in the delivery systems needed to transfer mRNA, which is inherently susceptible to both enzymatic and chemical degradation, into cells.
However, mRNA is still a new modality with increasing knowledge and developing global regulatory guidelines, and there is still little precedent in terms of marketed products and regulatory approvals to help guide developers through the challenges associated with manufacturing and establishing quality control strategies. This situation could adversely impact control strategies and timelines and, in turn, increase the cost of batch products.
To help alleviate some of these issues, BioPhorum gathered a team of industry subject matter experts to create a resource of potential critical quality attributes (CQAs) for products from the early stages of drug development to commercial manufacturing, enabling quick identification of relevant CQAs. The result is Defining the required critical quality attributes (CQAs) and phase requirements for mRNA/LNP product development and manufacture.
We asked two of the paper’s authors — Elaine Peters, director, analytical capabilities, cell and gene therapy at Roche, and Philipp Hoch, laboratory head, quality control, Bioanalytics at Rentschler Biopharma SE — some questions to explore the key areas in more detail.
Based on their combined industry experience, the authors provide not only recommendations of relevant CQAs but also guidance on the stage at which their testing may occur and the category of control strategy (e.g., release, stability, etc.), justifications and considerations for different mRNA product types, and suggested characterization information to inform mRNA developers’ quality control strategies.
1. The USP has released the second proposed version of its guidelines for mRNA vaccine quality (DS, now including some DP-related information). How do you see BioPhorum’s recommendations complementing this or any other available analytical/quality-related documents in this space?

Many other documents in the space have a vaccine focus. BioPhorum’s publication is indication-agnostic, covering attributes that apply to vaccines and therapeutics independent of indication. This is particularly timely as the field of mRNA products continues to rapidly expand.

2. What are the overall CQAs for mRNA/LNP product development and manufacture? And what are BioPhorum’s recommendations for mitigating or solving the challenges associated with them?
PETERS: The complete list of CQAs will always be product-specific; however, there are many common CQAs that apply to all products. Highlighting the attributes that a developer should consider, the rationale behind their importance, and potential methodologies to evaluate them is critical. Understanding this rationale will help developers establish their CQAs and have a starting point on which methods to use. This should help them mitigate some early challenges.
The team identified CQAs in five main categories: purity and product related impurities; safety tests; strength, identity, and potency; product quality and characteristics; and other obligatory CQAs. In each case they also provided suggested analytical methodologies to use and when to test.
Table 1: Summary of CQAs for mRNA/LNP product development and manufacture. The CQAs contained in the table may be considered as potential critical quality attributes ((p)CQAs), nCQA (non-critical quality attributes), or CQA (critical quality attributes) depending upon individual product, process knowledge, and regulatory/legislative requirements.
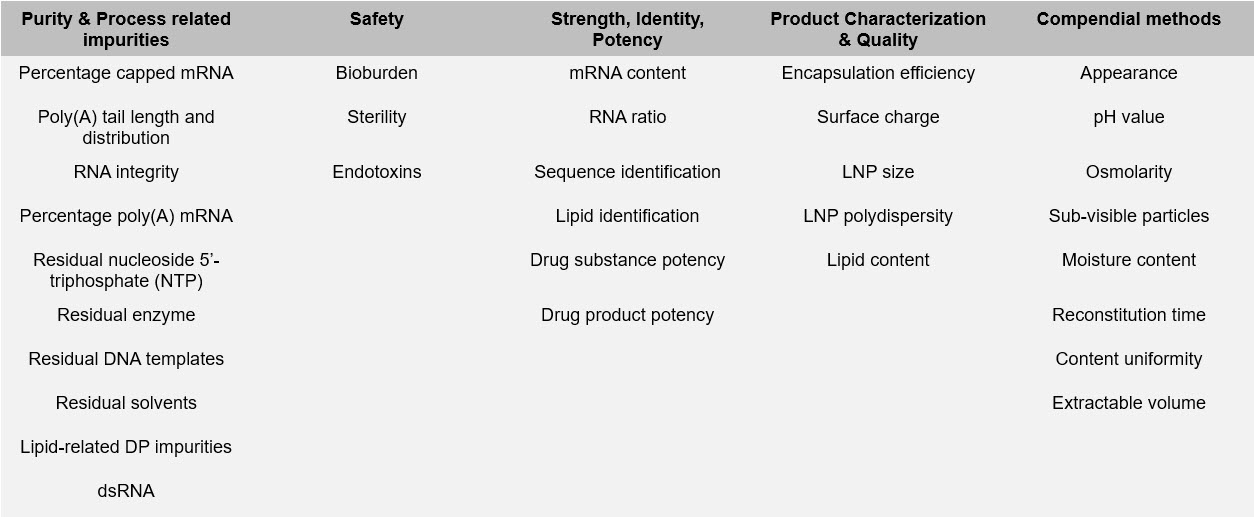
3. What are the nCQAs? And what are BioPhorum’s recommendations for mitigating or solving the challenges associated with them?
PETERS: Early product characterization is key. That is how you assess what your critical quality attributes are, which may differ from product to product. So, it is essential to start early in development, evaluate multiple potential CQAs (pCQAs) early on, and establish assays to identify actual CQAs and appropriate testing and specifications to support late-phase studies. Even if an attribute does not turn out to be “critical,” it can provide valuable information on your product and/or your process, so it is always worthwhile monitoring non-critical characteristics.
4. In addition to clinical data, what resources (e.g., standards, publications, regulatory guidance, etc.) will be the most valuable to clarify our understanding of these critical ranges (CQAs and CPPs)?
PETERS: Ultimately, CQAs and CPPs will always be defined for an individual product. Process and product development experience with these or similar products and characterization data is key. However, there is limited clinical experience with mRNA products, which means correlating CQAs to clinical outcomes is challenging. It would be good to have regulatory guidance. There are moves to address the standards/guidance gap. For example, the WHO has a document that is helpful for vaccines for infectious diseases and the USP recently issued draft guidance available for public consultation, but it is still early days. The success of COVID-19 vaccines tends to mask the fact that this is still a new area for drug development. As the field grows and evolves, I expect to see more formal resources from health authorities, etc.
5. What are the biggest gaps in analytical and/or clinical knowledge that continue to make our understanding of CQAs and critical process parameters (CPPs) “fuzzy”?
PETERS: For me, potency is a critical subject — both potency assays and understanding the definition of “potency” for an mRNA product. This is key to establishing and controlling CQAs and CPPs, as is understanding how they impact function and potency. Without a clear definition of what “potency/function” means for an mRNA product and assays to assess potency, it is challenging to fully understand and control your CQAs and CPPs.
A key question to ask during development is: What is the potency of an mRNA product? Is it only protein expression? Do you need to cover all aspects of function (e.g., transfection, translation, expression, and functionality of encoded sequence)? This is an area where a matrix approach for potency is ideal. Clear guidance and alignment on the definition of potency for an mRNA product between developers and regulatory agencies would be invaluable.
Protein-based active pharmaceutical ingredients have been around for a while and thus broad knowledge about analytical requirements and methods that are suitable to entirely characterize and analyze them is available. For mRNA, literature on several analytical methods is available, ranging from relatively simple techniques (such as agarose gel electrophoresis) to more sophisticated methods (such as next-generation sequencing). A huge variety of commercial analytical kits and solutions are also available.
HOCH: Unfortunately, there is limited knowledge on which methods, and combinations of them, are appropriate to build up a reasonable analytical grid that is suitable to cover all CQAs in terms of characterization, in-process controls, and release analytics without needing excessive effort and creating analytical redundancy.
6. How would you assess the current state of mRNA analytical development, particularly in terms of method availability, suitability, reliability, and/or reproducibility for mRNA therapies?
PETERS: Generally, analytical development for mRNA products is in good shape. mRNA concentration and mRNA integrity have well-established methodologies and there are some mRNA-specific tests where the field is gaining more experience, e.g., capping, double-stranded RNA (dsRNA), Poly(A) tail, and LNP encapsulation.
HOCH: There is a lot of literature on reliable, reproducible, and suitable analytical methods available for the analysis of mRNA, and several commercial kit solutions are on the market. Analytical development for mRNA analysis has been performed over a long period and before the COVID-19 pandemic brought mRNA therapies into focus.
7. To what extent are we capable of using and easily applying existing analytical technologies for mRNA therapies?
PETERS: Many of the required analytical tests and methods are applicable and can easily be used for mRNA products. These include, for example, the safety tests (sterility, endotoxin, bioburden) and general methods (e.g., appearance, pH, particulates, and osmolality). Other required testing also applies (e.g., identity, purity, strength, and potency), as well as removal of process impurities and animal-derived reagent testing. The difference is that while the concepts are the same as for other biologics, the specifics of how these are executed differ. One example is that mRNA products will have different process/product impurities than other types of products (e.g., dsRNA and residual nucleotides), but the requirement to have testing to show reduction/elimination is the same.
HOCH: I agree. Many of the available methods could be performed with common laboratory equipment, making them readily available, e.g., HPLC, capillary electrophoresis, (real-time) PCR cyclers, or microplate readers.
8. Where do you feel we may need better, more mRNA-appropriate analytical technologies?
HOCH: We don’t necessarily need more or better analytical methods, but more guidance is needed on the CQAs that should be considered at which stage and which methods might be the right ones to address these.
9. For which quality attributes do you see the greatest debates occurring in the mRNA/RNA space over the “best” method for measuring those specific quality attributes? Is there consensus in BioPhorum membership on the best method?
HOCH: The measurement of potency might be an important future matter for debate. It might be defined as the capability of mRNA to serve as a template for the translation of a protein, but at the same time the potency of an mRNA drug is always connected to the potency of the encoded protein and strongly affected by the cellular uptake and several other factors that are not intrinsic to the mRNA itself. A consensus on the “best” method of measurement appears to be elusive. To my knowledge, there currently is no consensus on how to address this parameter and whether the determination of potency may be derived from analytical controls of a set of other CQAs, such as sequence, integrity, capping, tailing, etc.
PETERS: In addition to potency, measuring dsRNA would be another hot topic. dsRNA can initiate an innate inflammatory response, activate mRNA sensors, or promote mRNA interference. It is important to remove as much dsRNA as possible from the final product. The current gold standard is an immunoblot (dot blot) test, but there is consensus in the field that this is not adequate and that we need better methods. The immunoblot provides only a high-level view of the quantity of dsRNA in a product; it does not tell you anything about the structure or type of dsRNA. Is it coming from complementary strands? Is it a “hairpin”? This information is critical to understanding its origin and how process development can reduce or eliminate it. You also need to understand the impact on function and safety. In general, a lot of the focus in the field is on the analytics and development of the mRNA itself, and the industry is starting to recognize the need for greater focus on attributes of the lipids/LNPs.
10. What are the key takeaways you’d like to impart?
HOCH AND PETERS: Expectations for CQA testing differ by product, making them difficult to align. Also, every company has different mRNA product development strategies depending upon several factors, such as indication. This article is a good starting point for others to consider, but each company must make its own decisions in line with regulators’ expectations.
HOCH: There are also multiple attributes and methods that can be used to measure various CQAs; this is not a one-size-fits-all approach. Nonetheless, some analytical methods can be used in a platform approach for different mRNA-based products, which might save future costs and effort.
Moving Forward
This Q&A discussion illustrates some of the issues facing mRNA developers while regulators continue to develop their guidelines. Ensuring consistent high-quality mRNA drug product manufacturing under continually compressed timelines remains a significant challenge for developers. By continuing collaborative discussions, we can support developers and help them identify relevant CQAs, the stage at which their testing may occur, and the category of control strategy (e.g., release, stability, etc.).
Acknowledgment
Special thanks to Christine Boswell of BioPhorum for her assistance with this Q&A.