
Trends In FDA FY 2025 Warning Letters
By Erin Hartmann and Liz Oestreich, ELIQUENT Life Sciences

The U.S. FDA issued a total of 303 warning letters (WLs) to drug and biologics products in Fiscal Year 2025 (FY25), a 59% increase from FY24 (190).1 Of those, 135 letters were based on an FDA inspection (“inspection-based warning letters”), including 16 letters issued to clinical investigators or sponsors following inspections conducted as part of the FDA’s Bioresearch Monitoring (BIMO) Program. The other 167 letters, the “non-inspection-based warning letters,” were issued pursuant to either a website, labeling, and/or promotional communications review (143 letters), a records request for remote evaluation of a facility under section 704(a)(4) of the FDCA (704(a)(4) request) (22 letters),2 or a failure to register and list a product (electronic drug registration and listing system violations, eDRLS) (two letters). The purpose of this article is to articulate key themes and trends of the FY25 warning letters for drug and biologic products. While we focus mainly on analyzing inspection-based warning letters, we also offer general context on all warning letters for these product categories.
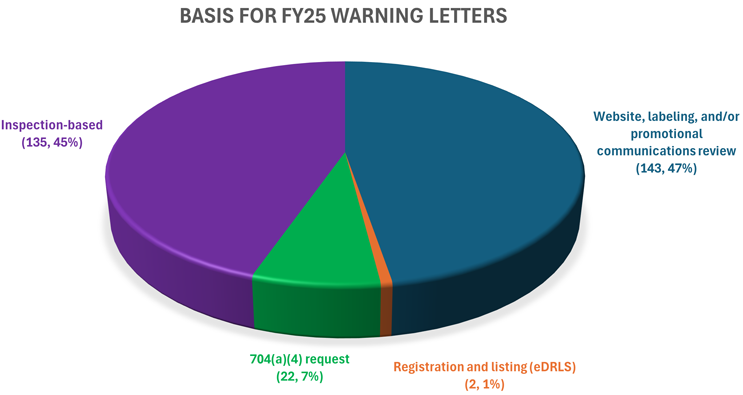
The increase in letters issued in FY25 is largely due to a surge of unapproved drugs flooding the market, many that were produced by compounders. FY25 inspection-based warning letters also surpassed the 111 inspection-based letters issued in FY24,3 as well as the 94 inspection-based letters issued in FY234 and the 74 inspection-based letters issued in FY22.5 This is an impressive feat given the significant reduction in force across the agency in the spring of 2025.6 The FY25 breakdown for warning letters (see graph above) varies greatly from previous years, with the majority of letters coming from a non-inspection-based review rather than an on-site inspection. For example, in FY24, 27% of warning letters were issued following a website, labeling, and/or promotional communications review, 59% were issued following an inspection, 10% were issued following a 704(a)(4) request, and 4% were issued following another type of review.7
Non-Inspection-Based Warning Letters
Two areas of regulatory focus in FY25 included unapproved glucagon-like peptide-1 (GLP-1) products and telehealth platforms8 selling compounded products. CDER also launched unapproved drug initiatives for ophthalmic products, drugs claiming to treat life-threatening conditions, products with hidden drug ingredients, and unauthorized sunscreen dosage forms.9
A significant number of WLs, 58 letters, were issued to online telehealth platforms selling compounded products. A total of 73 non-inspection-based warning letters were issued to facilities that produce weight loss products, and 55 of the letters were sent to producers of compounded products. The increased popularity, demand, and availability of GLP-1 products indicated for weight loss has dominated the market.
Another significant portion of WLs, 73, were issued to firms selling, distributing, and/or manufacturing unapproved new drug products. Nineteen of those WLs concern OTC products: eight letters were for unapproved sunscreen dosage forms,10 five letters were for unapproved ophthalmic products, and six letters were for unapproved external analgesics intended to be used prior to sensitive cosmetic procedures. Six of the 73 letters were issued by CBER for unapproved new biologic/drug products, with an emphasis on umbilical cord-derived products and exosome products. Additionally, the FDA continued its regulatory action against the sale/distribution of unapproved opioid products. Thirteen of the 73 letters were issued to firms selling unapproved opioids, benzodiazepines, and/or Schedule II stimulants on their websites. In those WLs, the agency notes the severity of the opioid epidemic as well as the strong potential for abuse of benzodiazepines and Schedule II stimulants. In total, the FDA highlighted the public health and safety concerns posed to consumers who use these firms’ unapproved products in 50 of these 73 WLs.
FDA also continued to utilize its authority under section 704(a)(4) of the FDCA in FY25. Twenty-two warning letters were issued following 704(a)(4) requests,11 13 of which were issued to firms located in China. Three were issued to firms in India, and two were issued to firms in Turkey. One letter each was issued to a firm in South Africa, Canada, and Australia, and only one letter was issued to a domestic firm. This suggests that the agency is using 704(a)(4) requests as a strategic tool for international facilities, likely for the purpose of utilizing resources efficiently. Notably, all but two of the warning letters issued following a 704(a)(4) request stated that the firm was placed on an import alert.12
Another key area of regulatory focus in FY25 compared to prior years includes the crackdown on promotional communications. The agency issued 11 WLs in FY25 following the review of promotional materials. Ten of the 11 WLs cited promotional materials for prescription products and found the promotional communications to be false and misleading, primarily on the grounds that the communications contained information about the efficacy of the product but failed to include sufficient information about the risks. Ten of the 11 letters also cite public health concerns resulting from the promotional communications, stating they create a misleading impression about the safety and efficacy of the product. Further, eight of the 11 firms that received a WL failed to submit the promotional materials under Form FDA-2253 at the time of initial dissemination. These warning letters were issued in line with the agency’s announcement in September 2025 that it would be cracking down on “deceptive drug advertising.”13
Finally, two letters were issued exclusively for failure to register and list; however, registration and listing failures were also noted in one warning letter following a 704(a)(4) request and in five inspection-based letters.14
Agency Focus
The reorganization of the FDA’s Office of Regulatory Affairs (ORA) into the Office of Inspections and Investigations (OII) was implemented on October 1, 2024. The goal of this reorganization was to allow OII to focus on inspections, investigations, and imports. It also triggered “unification of compliance functions within the centers,” leading to consolidation of all field compliance functions into the relevant product centers.15 This unification and consolidation streamlined the process for issuing warning letters and improved collaboration between the field and the centers. In December 2025, Melissa Mendoza, director of CBER’s Office of Compliance shared, “[t]he reorganization, among its many benefits, has led to streamlined compliance reviews. It has taken us, so far, one-third of the time to issue warning letters from the date that the inspection closes to the date of warning letter issuance.”16
The April 2025 reduction in force (RIF) at FDA does not appear to have impacted WLs for FY25. We will continue to monitor the potential impacts the RIFs may have in FY26 and beyond.
Inspection-Based Warning Letters
For the purpose of this article, we chose to analyze 135 of the inspection-based warning letters, removing one letter due to its focus on a medical device product.17
Over-the-counter (OTC) products continue to dominate as the largest product category, receiving 61 warning letters in FY25. These products mainly include topical analgesics, hand sanitizers, and sunscreens. Additional product categories include finished drug products (19), active pharmaceutical ingredients (APIs) (13), CBER-regulated products (13), compounded products (12), and one combination product. The remaining 16 letters were issued to various products in clinical trials.18
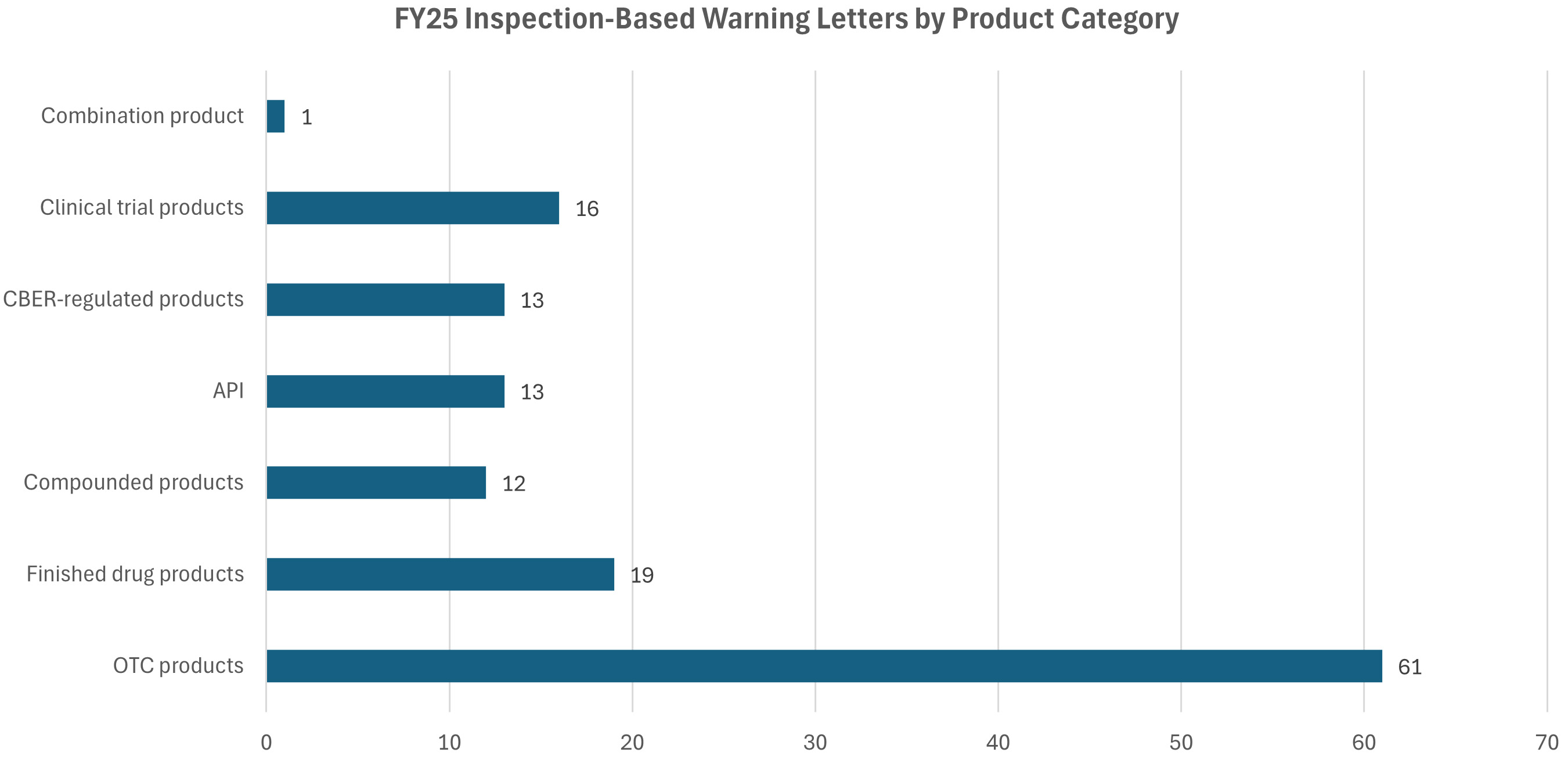
Click to Enlarge
Eighty-five inspection-based warning letters were sent to U.S. firms (~63%), while 50 (~37%) were sent to firms located outside of the United States. In FY24, roughly 61% of inspection-based warning letters were issued to domestic firms.19 FDA issued warning letters to firms in fewer countries in FY25 (13 different countries in FY25 vs. 19 different countries in FY24); however, the overall percentage of letters issued to domestic vs. foreign firms remained consistent (61% domestic in FY24; 63% domestic in FY25).20
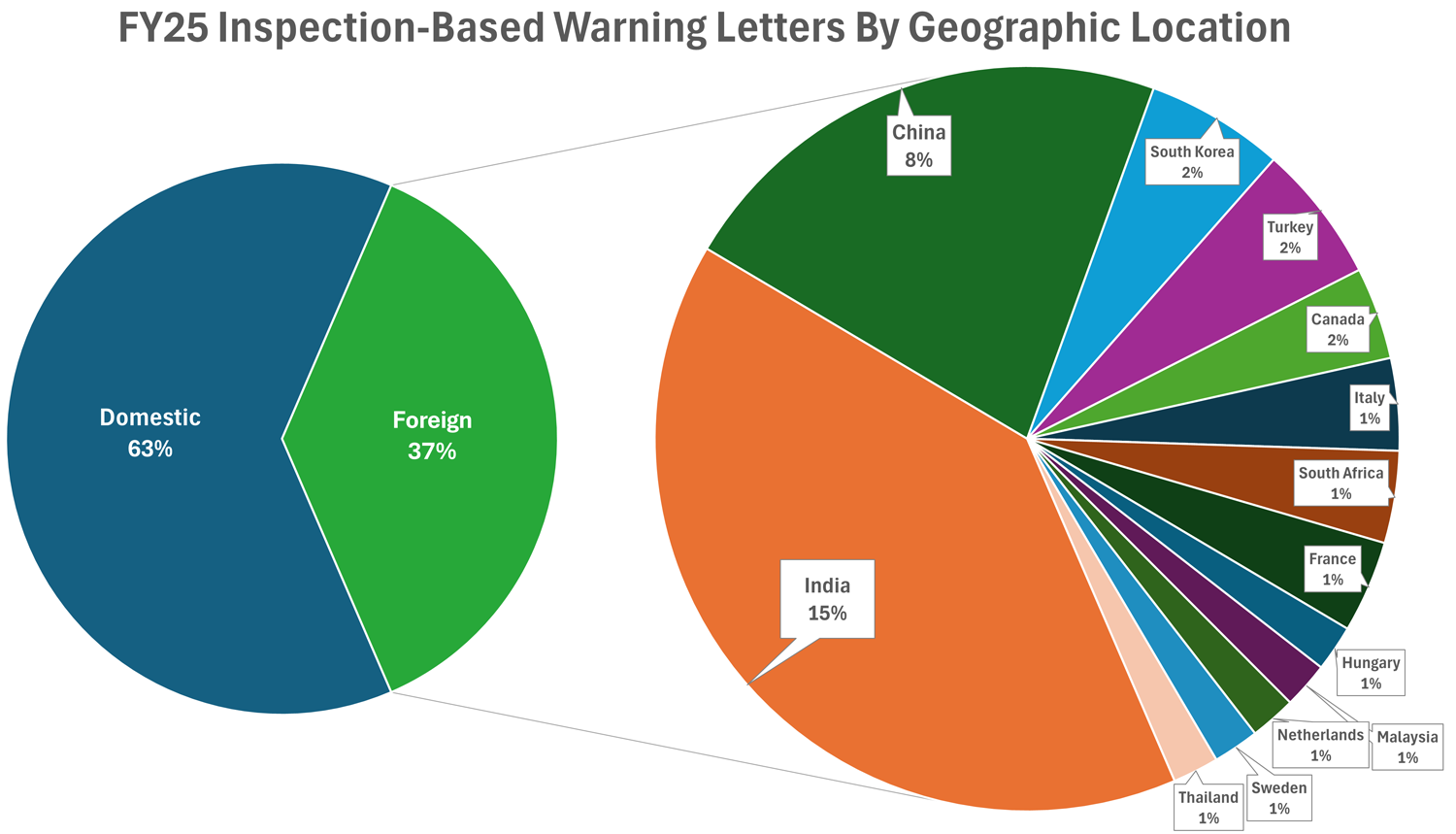
Click to Enlarge
Two Indian firms, one Chinese firm, and one Canadian firm refused entry or otherwise restricted FDA investigators from accessing necessary areas or documents during an inspection.22
Key Themes For Inspection-Based Warning Letters
Compounding
Two compounding pharmacies regulated under section 503A of the FDCA (503A pharmacies) and 10 outsourcing facilities regulated under section 503B of the FDCA (503B facilities) received warning letters in FY25. Six of the letters sent to outsourcing facilities include a recommendation to engage a third-party consultant with relevant sterile drug manufacturing expertise to assist with a comprehensive evaluation that includes review of aseptic processing techniques. One of the two letters sent to 503A pharmacies also includes this recommendation. Six of the 503B facilities that received warning letters also initiated voluntary recalls of product prior to receipt of the warning letter. Both letters sent to 503A pharmacies included citations for insanitary conditions. Given the sharp uptick in unapproved products (specifically compounded GLP-1 products), it is not surprising to see warning letters in this space. Under section 503B(b) of the FDCA, a compounder can register as an outsourcing facility with FDA. Drug products compounded by or under the direct supervision of a licensed pharmacist in an outsourcing facility qualify for exemptions from the drug approval requirements in section 505 of the FDCA [21 U.S.C. § 355(a)], the requirement in section 502(f)(1) of the FDCA [21 U.S.C. § 352(f)(1)] that labeling bear adequate directions for use, and the Drug Supply Chain Security Act requirements in section 582 of the FDCA [21 U.S.C. § 360eee-1] if the conditions in section 503B of the FDCA are met.23 A good example of a compounder’s failure to meet 503B requirements can be found in the warning letter issued to Empower Clinic Services, LLC dba Empower Pharma. In the warning letter, FDA includes several repeat observations from 2022, including discoloration on equipment in the ISO 5 area, monitoring environmental conditions in aseptic processing areas, and failure to establish and follow appropriate written procedures to prevent microbiological contamination.24
Cosmetics
On December 29, 2022, the Modernization of Cosmetics Regulation Act (MoCRA) was signed into law expanding FDA’s regulatory authority over cosmetic products. Since then, implementation regulations have been rolled out requiring cosmetic industry compliance with registration and listing, product labeling, safety substantiation, mandatory recall authority, adverse event reporting, and good manufacturing practice requirements. In FY25, 27 letters included language that “[a]ny cosmetics you manufacture must comply with applicable statutory and regulatory requirements, including the FD&C Act. A cosmetic is deemed adulterated under section 601(c) of the FD&C Act (21 U.S.C. 361(c)) if it has been prepared, packed, or held under insanitary conditions whereby it may have become contaminated with filth, or whereby it may have been rendered injurious to health.”25 The agency notes that some of the conditions that cause drug products to be adulterated may also cause cosmetic products to be adulterated. As the cosmetic regulated industry under MoCRA matures, we anticipate that violations of the MoCRA will be included in warning letters.
Recalls
Nineteen warning letters were accompanied by product recalls. Last year, we noted a new trend in warning letter language specifically concerning teleconferences between FDA and a firm regarding the initiation of a voluntary recall. The trend continued this year, with six of the 19 warning letters including language of this kind. For example, FDA writes in the Kenil Healthcare Private Limited warning letter: “On March 26, 2025, FDA held a teleconference with you and your U.S. Agent, JMM Services LLC. We recommended that you recall all your drug products within expiry currently in distribution to the U.S. market. On April 9, 2025, your firm committed to recall the products within expiry. After several subsequent attempts were made to contact you and your registered U.S. Agent regarding the recall, you initiated the recall of all your drug products on April 24, 2025.”26 FDA does not have mandatory recall authority over drug and biologic products. Thus, it relies on industry to initiate voluntary recalls to remove violative product from the market in a timely manner. The practice of detailing recall discussion during FDA teleconferences in warning letters is a strategy to encourage industry to take timely action over decisions that impact patients.
Top Citations
While the number of inspection-based warning letters has increased consistently over the last four years, there is no significant difference in top citations included in FY25 inspection-based warning letters compared to warning letters from the past four years.
The most frequent observation in FY25 was the failure of a quality control unit to ensure drug products manufactured are in compliance with current good manufacturing practices (cGMP), in accordance with 21 CFR 211.22. Forty-five letters cite 21 CFR 211.22, and 17 letters include a specific reference to subsections (a), (d), or both. A core function of a firm’s quality unit is to provide adequate oversight for the manufacture and release of products in compliance with cGMP.
Regulation 21 CFR 211.100(a) was cited 51 times in FY25. This citation highlights deficiencies in process validation and is another core concept in quality drug manufacturing. In its warning letter to Oasis Medical Inc., FDA explains, “Process validation evaluates the soundness of design and state of control of a process through its lifecycle. Each significant stage of a manufacturing process must be designed appropriately to ensure the quality of raw material inputs, materials, and finished drugs. Process qualification studies determine whether an initial state of control has been established. Successful process qualification studies are necessary before commercial distribution. Thereafter, ongoing vigilant oversight of process performance and product quality are necessary to ensure you maintain a stable manufacturing operation throughout the product lifecycle.”27
Regulations 21 CFR 211.84(d)(1) and 211.84(d)(2) remain a top citation in warning letters, and the agency continues to focus on risk of substitution or sub- or super-potent ingredients in OTC products like hand sanitizer. These observations note failure to appropriately verify the identity of each component and reliance on component suppliers’ test analyses without independent verification. This citation was included in letters for products such as hand sanitizer, hand soaps, and other topical drug products at high risk for diethylene glycol (DEG) or ethylene glycol (EG) contamination. Forty of the 48 letters cited both 211.84(d)(1) and 211.84(d)(2) while the remaining eight letters cited one or the other.
|
21 CFR Section |
Related WL Language |
# of WLs |
|
211.22: Responsibilities of quality unit |
Your firm’s quality control unit failed to exercise its responsibility to ensure drug products manufactured are in compliance with cGMP and meet established specifications for identity, strength, quality, and purity. |
62 |
|
211.100(a): Absence of written procedures |
Your firm failed to establish adequate written procedures for production and process control designed to assure that the drug products you manufacture have the identity, strength, quality, and purity they purport or are represented to possess. |
51 |
|
211.84(d)(1) and (d)(2): Identity testing and reports of analysis |
Your firm failed to conduct at least one test to verify the identity of each component of a drug product. Your firm also failed to validate and establish the reliability of your component supplier’s test analyses at appropriate intervals. |
48 |
|
211.192: Investigations |
Your firm failed to thoroughly investigate any unexplained discrepancy or failure of a batch or any of its components to meet any of its specifications, whether or not the batch has already been distributed. |
47 |
|
211.166: Stability testing |
Your firm failed to establish and follow an adequate written testing program designed to assess the stability characteristics of drug products and to use results of stability testing to determine appropriate storage conditions and expiration dates. |
40 |
Other notable citations include 211.165(a), (b), (d), or (e) related to testing and release for distribution, which is cited in 34 letters, and 211.160 for laboratory controls, which is cited in 33 letters.
Conclusion
As we see the changes of the OII reorganization take hold within the product centers, we can expect to see continued efficiency in the warning letter process. We applaud the agency for its continued work to protect the public amidst staffing changes and anticipate a continued focus on compounders, online retailers, and telehealth platforms in FY26.
- FDA issued a total of 303 warning letters issued in FY25 to drug and biologics manufacturers. However, one CBER-issued warning letter, O3UV, LLC (July 7, 2025), was for a CBER-regulated device, which we have excluded for the purposes of the analyses in this article.
- Remote regulatory assessments (RRAs) may be used by the FDA to support regulatory decisions and oversight activities; however, they are not considered inspections under sections 704(a)(1) (or 704(a)(5)) of the FDCA. See Conducting Remote Regulatory Assessments: Questions and Answers, Guidance for Industry (June 2025).
- Trends in FDA FY2024 Inspection-Based Warning Letters, March 7, 2025.
- Trends in FDA FY2023 Inspection-Based Warning Letters, February 13, 2024.
- Trends in FDA FY2022 Inspection-Based Warning Letters, February 21, 2023.
- Center for Drug Evaluation and Research & Center for Biologics Evaluation and Research Net Hiring Data (FY 2023-2027) (last updated Feb. 12, 2026).
- Trends in FDA FY2024 Inspection-Based Warning Letters, supra note 4. See also, Trends in FDA FY2022 Inspection-Based Warning Letters and Trends in FDA FY2023 Inspection-Based Warning Letters, supra notes 6 and 5.
- FDA listed warning letters issued to internet retailers/providers of various compounded drug under the term “Telehealth” on its webpage: Compounding: Inspections, Recalls, and other Actions.
- FDLI Enforcement and Litigation Conference, Jill Furman, Director Office of Compliance, CDER, December 2025.
- See FDA warning letter to Vacation Inc. (Aug. 6, 2025) was additionally uniquely cited for its unapproved sunscreen products being misbranded under section 502(i)(1) of the FD&C Act, 21 U.S.C. 352(i)(1) due to the packaging containers resembling food canisters customarily purchased by U.S. consumers. Specifically, the sunscreens “are presented in metal canisters with an ‘authentic tilt valve actuator’ that outputs a star-shaped foam and have a strong overall resemblance to the metal canisters ordinarily used to package whipped cream products and similar dessert toppings.” The agency highlights that “[p]ackaging drug products in containers that resemble food containers commonly used by adults and children can mislead consumers into mistaking the products for food, which is of particular concern as this increases the risk of accidental ingestion.”
- FDA’s warning letter to Cellebration LLC (Sept. 12, 2025) also included mention of FDA’s website review.
- Zhejiang Easyclean Daily Chemical Co., a Chinese firm, received a warning letter on May 14, 2025, following a 704(a)(4) request but was not placed on import alert. Cellebration LLC is a domestic firm located in Puerto Rico and thus was not placed on import alert.
- See FDA News Release: FDA Launches Crackdown on Deceptive Drug Advertising (September 9, 2025).
- See FDA non-inspection-based WLs: Prodose, Inc. (Mar. 28, 2025), Shenzhen Hengkaifeng Commerce and Trade Co., Ltd (May 6, 2025), Omni Lense Pvt. Ltd. (Oct. 2, 2024) (704(a)(4) request); FDA inspection-based WLs: Diora Kimya Sanayi ve Ticaret Limited Sirketi (Oct. 17, 2024), Choice All Natural, Inc. dba Om Botanical (Oct. 21, 2024), Dr. Brite, LLC (Nov. 21, 2024), Jagsonpal Pharmaceuticals Limited (Feb. 5, 2025), and Medical Chemical Corporation (July 9, 2025).
- Jill Furman, FDLI Enforcement Conference (December 2025).
- Melissa Mendoza, FDLI Enforcement Conference (December 2025).
- FDA’s warning letter to O3UV, LLC (July 7, 2025) regarding autohemotherapy devices was not included in this article. See supra note 1.
- Note that for purposes of this article, firms were included in the OTC product category if the warning letter mentioned an OTC product. However, this does not exclude the possibility that the firm also produces prescription drug products and/or other finished drug products. Similarly, firms listed under the finished drug products category may also produce OTC products, but an OTC product was not specifically mentioned in the warning letter.
- Trends in FDA FY2024 Inspection-Based Warning Letters, supra note 3.
- Id.
- For the purpose of this analysis, we categorized the Sanofi warning letter (Jan. 15, 2025) as a domestic inspection since the Genzyme facility is located in New York State, not in France.
- See FDA warning letters to Jagsonpal Pharmaceuticals Limited (Feb. 5, 2025) (India), Tianjin Darentang Jingwanhong Pharmaceuticals Co. Ltd. (Oct. 30, 2024) (China), Unexo Lifesciences, Private Limited (Nov. 6, 2024) (India), and Brands International Corporation (Dec. 17, 2024) (Canada).
- FDA warning letter to Empower Clinic Services, LLC dba Empower Pharma (April 2, 2025).
- Id.
- See, e.g., FDA warning letter to Eurosirel S.P.A. (Nov. 20, 2024).
- FDA warning letter to Kenil Healthcare Private Limited (June 12, 2025).
- FDA warning letter to Oasis Medical Inc. (July 15, 2025).
About The Authors:
 Liz Oestreich, J.D., is senior vice president of regulatory compliance at Eliquent Life Sciences (formerly Greenleaf Health). She brings more than 10 years of regulatory experience and a diverse background of legal, public policy, and non-profit sector knowledge to her position. As a consultant, Oestreich provides strategic guidance on premarket and postmarket issues to drug, medical device, tobacco, and cannabis companies. She earned her J.D. from the University of the District of Columbia, David A. Clarke School of Law, and her B.A. from the University of Arizona.
Liz Oestreich, J.D., is senior vice president of regulatory compliance at Eliquent Life Sciences (formerly Greenleaf Health). She brings more than 10 years of regulatory experience and a diverse background of legal, public policy, and non-profit sector knowledge to her position. As a consultant, Oestreich provides strategic guidance on premarket and postmarket issues to drug, medical device, tobacco, and cannabis companies. She earned her J.D. from the University of the District of Columbia, David A. Clarke School of Law, and her B.A. from the University of Arizona.
 Erin Hartmann, J.D., is manager of regulatory affairs in Eliquent Life Sciences’ Quality and Compliance Practice. She analyzes FDA regulatory and compliance trends and provides strategic insights to clients. She is a graduate of the University of Texas School of Law and has a B.S. in biology from Case Western Reserve University.
Erin Hartmann, J.D., is manager of regulatory affairs in Eliquent Life Sciences’ Quality and Compliance Practice. She analyzes FDA regulatory and compliance trends and provides strategic insights to clients. She is a graduate of the University of Texas School of Law and has a B.S. in biology from Case Western Reserve University.