
The Shirokizawa Matrix: Determining The Level Of Effort, Formality, & Documentation In Cleaning Validation
By Andrew Walsh, Thomas Altmann, Ralph Basile, Joel Bercu, Ph.D., Alfredo Canhoto, Ph.D., David G. Dolan, Ph.D., Pernille Damkjaer, Andreas Flueckiger, M.D., Igor Gorsky, Jessica Graham, Ph.D., Ester Lovsin Barle, Ph.D., Ovais Mohammad, Mariann Neverovitch, Siegfried Schmitt, Ph.D., and Osamu Shirokizawa
 Part of the Cleaning Validation For The 21st Century Series
Part of the Cleaning Validation For The 21st Century Series
The International Congress on Harmonization Quality Risk Management Guidance (ICH Q9) lists both cleaning (in Annex II.4) and validation (in Annex II.6) as potential areas for the application of quality risk management.1 This clearly implies that the ICH Q9 principle for adjusting the level of "effort, formality, and documentation" based on the level of risk could be applied to cleaning and its validation. Previous articles discussed how science-based and data-derived scales could be created from HBELs (health-based exposure limits), from the process capability (Cpu) of cleaning processes, from the detection limits for total organic carbon (TOC) analyses of these compounds, or from visual inspection.2-5 Another article discussed how these scales could be used to measure the level of risk in cleaning validation.6 This article builds on these discussions and shows how these HBEL-based and process capability-based scales can be combined into a matrix that provides a clear visual guide for adjusting the level of effort, formality, and documentation for cleaning validation based on the level of risk.
Quality Risk Management Under ICH Q9
As previously discussed,6 ICH Q9 describes two primary principles of quality risk management (QRM):
• "The evaluation of the risk to quality should be based on scientific knowledge and ultimately link to the protection of the patient; and
• The level of effort, formality and documentation of the quality risk management process should be commensurate with the level of risk".
From these two primary principles, it can be understood that if we can determine the level of risk to a patient from cleaning, then the level of cleaning validation effort, its formality, and its documentation could be adjusted accordingly. Stated simply, cleaning validation efforts for low-risk products should not require the same level of effort as for high-risk products. This is perfectly logical. The level of effort, formality, and documentation for cleaning validation should correspond to the level of risk, which includes the available knowledge of a cleaning process and the nature of the product.
ICH Q9 defines risk (in general) as:
"It is commonly understood that risk is defined as the combination of the probability of occurrence of harm and the severity of that harm".
and further on:
"...the ability to detect the harm (detectability) also factors in the estimation of risk".
We also see risk can be more formally expressed as:
Risk = f (Severity of Hazard, Level of Exposure to Hazard, Detectability of Hazard)
Now, if the hazard is intrinsic to an active pharmaceutical ingredient (API) and the risk being considered is harm to a patient from exposure to residues of that API after cleaning, then this equation can be further refined to:
Cleaning Risk = f (ToxicityAPI residue, Level of ExposureAPI residue, DetectabilityAPI residue)
So, if we can measure these parameters of toxicity, exposure, and detectability we should be able to locate our position on the continuum shown in Figure 1.
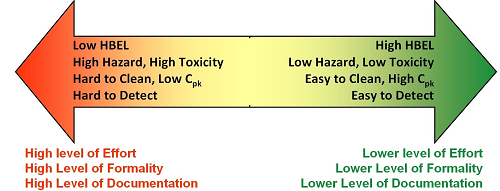
Figure 1: Continuum of risk in cleaning based on hazard, exposure, and detectability
Science- And Risk-Based Scales For Toxicity, Exposure, And Detectability
The scales presented in the preceding four articles2-5 offer scientifically based methods to measure these risk parameters using actual data. Since the scales presented in the four articles are all based on science and derived from actual data, they would consequently make good choices for evaluating the risk in cleaning, including patient safety.
These scales were originally developed as replacements for the typical scales used in failure modes and effects analysis (FMEA). The subjectivity of the scales typically used in FMEAs, and the lack of a scientific/statistical basis for their risk priority numbers (RPNs), often makes both the scales and their RPNs inappropriate for use in the pharmaceutical industry, as discussed in an earlier article.6 If pharmaceutical manufacturing is to advance to a science- and risk-based approach, the scales for severity, occurrence, and detectability used in FMEAs must be scientifically justified using science, process knowledge, and statistics. Such scales should be derived from, and based on, empirical data. Data such as this exists for cleaning and is readily available in pharmaceutical manufacturing production. As stated in the introduction, scales already exist that could be used for the following criteria:
- HBEL-derived Toxicity Scale → Severity of Process Residues2
- Process Capability-derived Scale → Occurrence of Exposure to Process Residues3
- TOC Detectability Index → Detectability of Process Residues 4
- Visual Detectability Index → Detectability of Process Residues 5
For example, in a cleaning process, if a failure mode could result in residues of an API remaining on equipment, then the HBEL-derived toxicity score of that API could be used as a severity score. Furthermore, if the process capability of the cleaning process is known, then its process capability-derived score could be used as an occurrence score (as the effectiveness of the cleaning process and the probability of residues remaining are known). Finally, if either the visual detectability index4 or the TOC detectability index5 is known, then one or both of these could be used as a detectability score. Since these scores are derived directly from empirical data, their values are specific and objective and should not be subject to debate, as happens frequently with traditional FMEA scales. It should be noted that detectability indexes may also be compiled from other available analytical test methods used, such as UV, HPLC, etc.
As a refresher on the previous articles,2-5 the HBEL-derived Toxicity Scale2 is based on converting HBEL (ADE/PDE) values to a scale in a manner similar to that used to create the pH (log-based) scale. By converting the HBEL value into grams and taking its negative logarithm, a continuous scale from 0 to 10 (as is typical of the severity scales used in FMEAs) can be generated for HBELs, ranging from a high value of 1 gram/day (low hazard) to a low value of equal to or less than 1 nanogram/day (high hazard).
The Process Capability-derived Scale3 depicts the process capability (Cpu) by converting actual cleaning data into a scale from 1 to 10 by taking the reciprocal of the Cpu and multiplying by 10. This results in a scale that has a high value (i.e., 10) associated with a high probability of failure and a low value (i.e., 1) associated with a low probability of failure, as is typical of an occurrence scale used in FMEAs. Note: The process capability for a Six Sigma cleaning process (Cpu = 2) was chosen as the midpoint of this scale.
The Toxicity/Cleaning Capability Matrix
Figure 2 shows an example of how the Toxicity Scale and Process Capability Scale can be combined into a matrix and groupings created based on the risk. This is called the Shirokizawa Matrix, and it can be used as a guide to determine the level of effort, formality, and documentation necessary for cleaning validation activities.

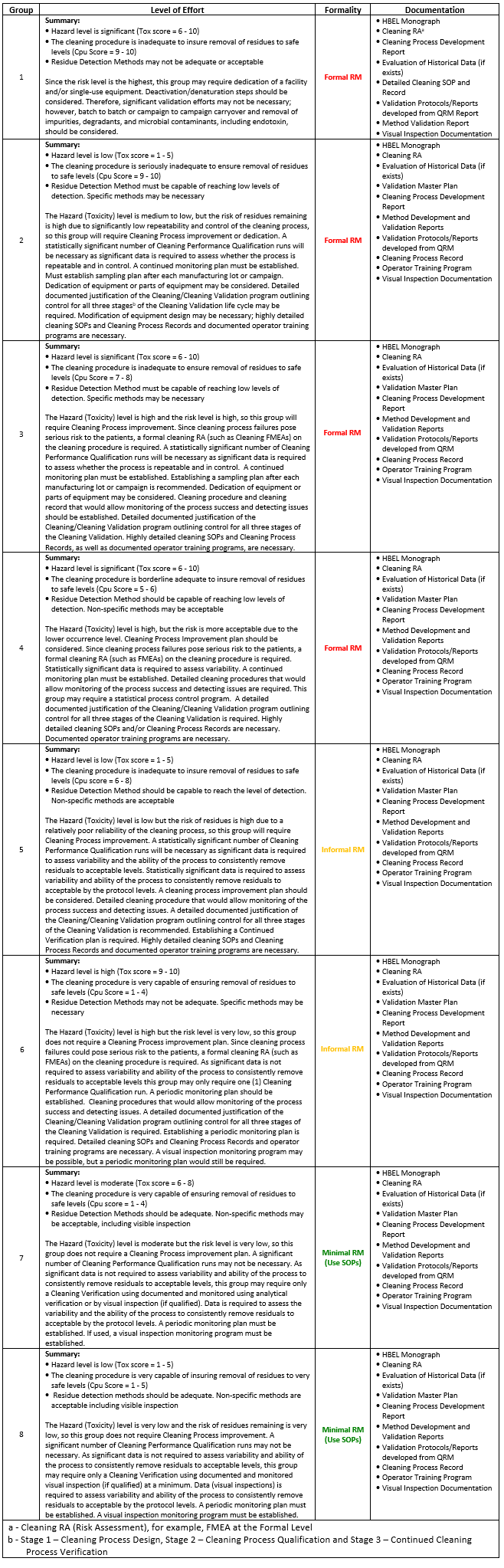
Figure 2: Shirokizawa Matrix of Cleaning Validation Effort – This matrix is an example and a potential model of how toxicity and process capability scores can be used together to determine an acceptable level of effort, formality, and documentation in cleaning validation programs.
The matrix in Figure 2 is separated into eight groups based on the coordinate positions in the matrix from the toxicity and process capability scores. The upper left quadrant contains the highest risk situations: high hazard compounds with poor cleaning processes. Compounds whose scores fall into this quadrant would require the highest levels of effort, formality, and documentation in their cleaning validation programs. The lower right quadrant contains the lowest risk situations: low hazard compounds with excellent cleaning processes. Compounds whose scores fall into this quadrant would require lower levels of effort, formality, and documentation in their cleaning validation programs.
Table 1 provides more detailed descriptions of the levels of effort, formality, and documentation that might be required for those compounds whose toxicity and their cleaning process capability positioned them in a particular group.
Table 1: Suggested Levels of Effort, Formality, and Documentation Required for Products Whose Toxicity and Process Capability Scores Place Them in One of the Eight Groups
Discussion
When the Risk-MaPP Guide7 first introduced the acceptable daily exposure (ADE) concept in 2010, one of the first reactions was that all the cleaning limits would become so low that companies would not be able to pass any cleaning validations. This concern was not based on any actual knowledge and was addressed by an article published in 2016 that evaluated 304 pharmaceutical products and compared their ADEs to 1/1,000th of their therapeutic dose limits.8 This article showed that in 85 percent of these cases, the limits were higher, even substantially higher, not lower, so this concern was unfounded. The next concern that was raised was that these higher HBELs would allow companies to "relax their cleaning efforts." This is another misguided assumption.
An original goal of Risk-MaPP, and subsequently of the ASTM E3106 Standard Guide,9 has been to implement the ICH Q9 principle that the "level of effort, formality, and documentation" of the cleaning validation process should be based on science and risk. A quick examination of the matrix in Figure 2 and the details in Table 1 will reveal that any "relaxation" of cleaning effort will lead to poor cleaning process capability and result in an increase of validation efforts. Only very good cleaning efforts can allow a company to reduce the levels of validation effort — even to the possibility of only a visual inspection program. The EMA indicated in Annex 15 that visual inspection could be used alone in certain cases10 and recently provided guidance on how a visual inspection program can replace analytical testing for release of equipment after cleaning.11 Such programs could provide significant operational benefits to companies that can successfully implement them. But the benefits for companies moving to the HBELs can only be realized if their cleaning processes are shown to be effective, repeatable, and safe.
Readers should understand that the groupings shown here are our initial recommendations and should undergo updating as experience is obtained using them. Further, their boundaries should not be considered rigid or fixed. For example, scores that put the risk at the intersection of 5 and 6 on the toxicity scale and 8 and 9 on the process capability scale could equally fall into groups 1, 2, 3, or 5. These groupings and their levels of effort should not be applied blindly, and serious consideration should be put into what efforts are truly necessary for each particular situation. These groupings are only meant to help guide the decision process. Therefore, practitioners are urged to exercise significant QRM efforts in which all elements of the firm’s cleaning and cleaning validation programs are carefully identified, analyzed, and evaluated.
The previous articles cited2-6 show how the application of scientific principles and statistical tools can be used to measure the level of risk in cleaning. Moving into the future, there should be a shift in the focus of cleaning validation programs to the derivation of HBELs and the development of cleaning risk assessments. The HBELs and the cleaning risk assessments will be used to inform the master plans and protocols for the level of cleaning validation that is necessary (Figure 3). The practice of simply using master plans and protocols from previous validations as templates should come to an end. The contents of master plans and protocols should be determined based on science and risk analysis and not on what was done the "last time."

Figure 3: The Cleaning Quality Risk Management Process – The activities in the BLUE zone are where full cleaning QRM efforts are applied. When these cleaning QRM efforts are completed, and the risk is now acceptable, the process can move into the GREEN zone. The GREEN zone is where the cleaning QRM level of effort, formality, and documentation can be reduced based on the results of the cleaning QRM efforts in the BLUE zone. This is where the grouping criteria from the Shirokizawa Matrix are applied.
Level Of Effort, Formality, And Documentation
The three concepts of "effort, formality, and documentation" may appear to be separate ideas, but in practice they are closely connected. For example, it is hard to imagine how a process that requires very little effort with very little documentation could have a very high level of formality or how a process that requires a very high level of effort with a very high level of formality can have very little documentation. Clearly, these aspects scale up or down with each other. Therefore, discussing them separately is not really possible or even useful. It is more informative to look at them together at each step of the risk management process (Figure 3).
The most important documentation in the cleaning validation process is found at the risk (hazard) identification stage, and these are the HBEL monographs for chemical hazards. At this stage, the levels of effort, formality, and documentation must remain at their highest, since all subsequent activities, decisions, and calculations flow from the information found in the HBEL monographs. There can be no "shortcuts" in the derivation of HBELs. An ASTM Standard Guide is currently being finalized on the derivation of HBELs12 that will be of immense help for companies at this stage.
The next most important documentation is the cleaning risk assessment. With the HBEL monograph in hand, the risk assessment process can begin. While the ASTM E3106 standard considers the HBEL at the risk (hazard) identification stage, microbiological hazards, equipment design hazards, and procedural hazards must also be considered. For a new product and a new facility, these other hazards may not be well understood, so a full risk (hazard) identification may be necessary. This may trigger risk reduction activities (Figure 3). On the other hand, for an established product in an established facility, many of these other hazards may have already been identified, considered, and mitigated. This prior knowledge should be leveraged and the level of effort during the risk (hazard) identification may be reduced. The level of effort, formality, and documentation at this stage will be high, but it can clearly be adjusted.
At the risk analysis stage, there may be no historical data to consider and analyze for a new HBEL in a new facility. This may require collecting substantial cleaning data to satisfy concerns about the level of risk. Cleaning process development studies are required and may be extensive. The level of effort, formality, and documentation may remain high. The lack of historical knowledge may likely trigger significant risk reduction activities. Alternatively, there may be substantial historical data on the cleaning process and an analysis may show that there is very little risk of cross contamination from this new product and, depending on the analyzed risk, only one verification run or even a simple visual inspection may be all that is necessary. A single cleanability test may be all that is needed to confirm that the cleaning process can be considered adequate and reliable.13 Consequently, the subsequent levels of effort, formality, and documentation may be reduced.
At the risk evaluation stage, where there is no historical data to consider and analyze, the master plan and protocols may require the collection of substantial data and performance of several cleaning performance qualification runs. Alternatively, if there is substantial historical data and the risk analysis showed that there was little risk of cross contamination from this new product, the protocol (or SOP) may require only one verification run or even just a visual inspection. Such an approach is very appropriate for the cosmetics and personal care industries, where there are many low-hazard compounds that could also be demonstrated to be low risk.
At the risk control stage, based on the preceding risk analysis and risk evaluations, continued monitoring using swab sampling after every changeover may be required, and if the remaining risk is considerably high, then equipment may be released only after acceptable test results. Or, the preceding risk analysis and risk evaluations may indicate that just visual inspection is adequate.
Finally, in the risk review stage, when a new product is being introduced, the level of effort, formality, and documentation should follow from the knowledge and understanding from the preceding risk analysis, risk evaluation, and risk control activities. This is an important subject, and a more detailed article on the introduction of new products is currently in development that will provide a flow chart for how a new product should be introduced within a cleaning QRM program.
One final observation: When it comes to “formality,” ICH Q9 does not go into any details about how this factor is affected by a risk assessment; in fact, the word "formality" is only used twice in ICH Q9 and both are simply mentioned. Therefore, ICH Q9 provides no real guidance on this aspect. The term "formality" has many definitions and uses, but for the pharmaceutical and related industries, it can be defined as "an established procedure or set of specific activities which need to be followed." Many companies already have detailed procedures for their cleaning validation programs, such as formal protocol templates, and they may wish to maintain these as they are. However, companies with lower-risk products and operations may want to simplify their cleaning validation process and move to SOPs and checklists or cleaning records. While there is no risk to patient safety from a company maintaining a high level of formality, there is the operational risk of performing excessive and unnecessary work and being slow to introduce new products. The higher the risk, the higher the level of formality, and the lower the risk, the lower the level of formality. Each company will need to decide for itself what level of formality to apply.
Conclusion
One of the goals of the ASTM E3106-18 Standard Guide was to provide a framework for a scientific risk- and statistics-based approach to cleaning processes and validation within an ICH Q9 framework and based on the FDA's 2011 Process Validation Guidance. The benefit of such an approach would be the ability to scale the level of "effort, formality, and documentation" of the cleaning validation process commensurate with the level of risk. The current ability to measure risk in cleaning provides an objective tool to focus cleaning validation efforts on the risks that are the most significant, based on the science behind the HBEL.
The authors know that most industry workers would agree that cleaning validation efforts for low-risk products (e.g., low toxicity, demonstrably easy to clean) should not require the same level of effort as for high-risk products (e.g., high toxicity, hard to clean). At the same time, we recognize that most industry workers prefer specific guidance on what they need to do, and simply stating "based on the level of risk" is not helpful or even useable. We have shown previously how the level of risk can be measured,6 but the question of what should be done at a particular level of risk still needed to be answered. The Shirokizawa Matrix described in this article is our first attempt at providing specific guidance on what levels of "effort, formality, and documentation" could be used. In this article we provide a science-based and data-driven approach to guide the level of effort, formality, and documentation for the cleaning of many healthcare products, including pharmaceuticals, biopharmaceuticals, nutraceuticals, cosmetics, and medical devices.
Peer Review:
The authors wish to thank our peer reviewers: Bharat Agrawal, James Bergum, Ph.D., Sarra Boujelben, Gabriela Cruz, Ph.D., Mallory DeGennaro, Parth Desai, Kenneth Farrugia, Angela Garey, Laurence O'Leary, Tri Chanh Nguyen, Miquel Romero Obon, Prakash Patel, Stephen Spiegelberg, Ph.D., Basundhara Sthapit Ph.D., and Joel Young for reviewing this article and for providing many insightful comments and helpful suggestions.
References:
- International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use, ICH Harmonized Tripartite Guideline, Quality Risk Management – Q9, Step 5, 9 September 2015, http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q9/Step5/Q9_Guideline.pdf.
- Walsh, Andrew, Ester Lovsin Barle, Michel Crevoisier, David G. Dolan, Andreas Flueckiger, Mohammad Ovais, Osamu Shirokizawa, and Kelly Waldron. "An ADE-Derived Scale For Assessing Product Cross-Contamination Risk In Shared Facilities" Pharmaceutical Online May 2017
- Walsh, Andrew, Ester Lovsin Barle, David G. Dolan, Andreas Flueckiger, Igor Gorsky, Robert Kowal, Mohammad Ovais, Osamu Shirokizawa, and Kelly Waldron. "A Process Capability-Derived Scale For Assessing Product Cross-Contamination Risk In Shared Facilities" Pharmaceutical Online August 2017
- Walsh, Andrew, Thomas Altmann, Alfredo Canhoto, Ester Lovsin Barle, David G. Dolan, Andreas Flueckiger, M.D., Igor Gorsky, Robert Kowal, Mariann Neverovitch, Mohammad Ovais, Osamu Shirokizawa and Kelly Waldron. "A Swab Limit-Derived Scale For Assessing The Detectability Of Total Organic Carbon Analysis" Pharmaceutical Online January 2018
- Walsh, Andrew, Thomas Altmann, Alfredo Canhoto, Ester Lovsin Barle, David G. Dolan, Mariann Neverovitch, Mohammad Ovais, Osamu Shirokizawa and Kelly Waldron. "An MSSR-derived Scale for Assessing the Detectability of Compound-Carryover in Shared Facilities" Pharmaceutical Online December 2017
- Walsh, Andrew, Thomas Altmann, Alfredo Canhoto, Ester Lovsin Barle, David G. Dolan, Andreas Flueckiger, Igor Gorsky, Jessica Graham, Ph.D., Robert Kowal, Mariann Neverovitch, Mohammad Ovais, Osamu Shirokizawa and Kelly Waldron "Measuring Risk in Cleaning: Cleaning FMEA and the Cleaning Risk Dashboard" Pharmaceutical Online April 2018
- ISPE Baseline® Guide: Risk-Based Manufacture of Pharmaceutical Products: A Guide to Managing Risks Associated with Cross-Contamination. (ISPE, Tampa, FL, First Edition, 2010), Vol. 7.
- Walsh, Andrew, Michel Crevoisier, Ester Lovsin Barle, Andreas Flueckiger, David G. Dolan, Mohammad Ovais (2016) "Cleaning Limits—Why the 10-ppm and 0.001-Dose Criteria Should be Abandoned, Part II," Pharmaceutical Technology 40 (8)
- American Society for Testing and Materials E3106-18 Standard Guide for Science-Based and Risk-Based Cleaning Process Development and Validation, www.astm.org.
- EudraLex, "Volume 4 – Guidelines for Good Manufacturing Practices for Medicinal Products for Human and Veterinary Use, Annex 15: Qualification and Validation," https://ec.europa.eu/health/documents/eudralex/vol-4_en.
- European Medicines Agency: Questions and answers on implementation of risk-based prevention of cross-contamination in production and “Guideline on setting health-based exposure limits for use in risk identification in the manufacture of different medicinal products in shared facilities," 19 April 2018 EMA/CHMP/CVMP/SWP/246844/2018
- American Society for Testing and Materials Work Item WK59975 - "Standard Guide for the Derivation of Health Based Exposure Limits (HBELs)," www.astm.org.
- Song, Ruijin, Alfredo Canhoto, Ph.D., and Andrew Walsh, "Cleaning Process Development: Cleanability Testing and "Hardest-To-Clean" Pharmaceutical Products" Pharmaceutical Online January 2019