
The 10 Most-Cited MHRA GMP Inspection Deficiencies By Annex/Chapter
By Barbara Unger, Unger Consulting Inc.
This is the second article in a two-part series reviewing the MHRA’s publication of the GMP deficiencies for drug product issued during inspections in 2018 and published in October 2019. Part 1 provided a high-level overview of the 2018 data and included additional trends from the two most recent MHRA reports (2015 and 2016). It also identified and evaluated the critical and major deficiencies from 2018.
product issued during inspections in 2018 and published in October 2019. Part 1 provided a high-level overview of the 2018 data and included additional trends from the two most recent MHRA reports (2015 and 2016). It also identified and evaluated the critical and major deficiencies from 2018.
In Part 2, we take a granular look at individual chapters and annexes identified in Part 1 and how the deficiencies are divided among the various paragraphs. We address Chapters 1, 3, 4 and 5, 6, 7, 8 and Annexes 1, 11, and 15. These are provided in order of decreasing number of deficiencies (see Figure 3 in Part 1). We provide the top 10 citations for each, or more in the two instances where there are ties for this honor. Each figure is followed by a table enumerating short versions of text of the top citations. The previous reports from the MHRA in 2015 and 2016 include text from actual inspections as examples, but that is not provided this year.
Chapter 1, Pharmaceutical Quality System
Figure 1 shows the top 10 citations from Chapter 1, and Table 1 provides the short text of the requirements, along with the number of times each was cited. The top two citations address problems with investigations and root cause analysis, which is not a surprise. This topic is among the most frequently cited FDA inspection observations. These two citations constitute 48 percent among the top 10 citing Chapter 1 and 29 percent of the total number of deficiencies citing Chapter 1. Also notable are the two citations associated with management oversight, paragraphs 5 and 6, which together constitute 12 percent of the total number of the top 10 and 7 percent of all deficiencies citing Chapter 1.
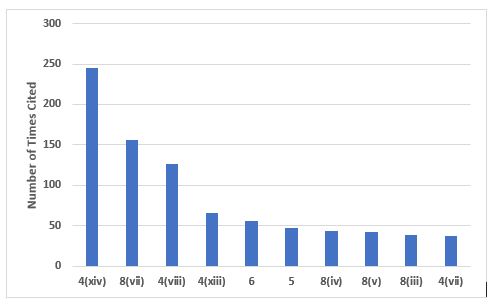
Figure 1: Top 10 deficiencies citing Chapter 1
Table 1: Top 10 Deficiencies Citing Chapter 1
|
Paragraph |
# |
Short Text |
|
1.4(xiv) |
245 |
An appropriate level of root cause analysis should be applied during the investigation of deviations, suspected product defects, and other problems. This can be determined using Quality Risk Management principles… |
|
1.8(vii) |
156 |
Any significant deviations are fully recorded, investigated with the objective of determining the root cause, and appropriate corrective and preventive action implemented; |
|
1.4(viii) |
126 |
A state of control is established and maintained by developing and using effective monitoring and control systems for process performance and product quality. |
|
1.4(xiii) |
66 |
After implementation of any change, an evaluation is undertaken to confirm the quality objectives were achieved and that there was no unintended deleterious impact on product quality; |
|
1.6 |
56 |
There should be periodic management review, with the involvement of senior management, of the operation of the Pharmaceutical Quality System to identify opportunities for continual improvement of products, processes, and the system itself. |
|
1.5 |
47 |
Senior management has the ultimate responsibility to ensure an effective Pharmaceutical Quality System is in place, adequately resourced, and that roles, responsibilities, and authorities are defined, communicated, and implemented throughout the organisation. |
|
1.8(iv) |
44 |
Instructions and procedures are written in an instructional form in clear and unambiguous language, specifically applicable to the facilities provided; |
|
1.8(v) |
42 |
Procedures are carried out correctly and operators are trained to do so; |
|
1.8(iii) |
38 |
All necessary facilities for GMP are provided, including: • Appropriately qualified and trained personnel; • Adequate premises and space; • Suitable equipment and services; • Correct materials, containers, and labels; • Approved procedures and instructions, in accordance with the Pharmaceutical Quality System; • Suitable storage and transport; |
|
1.4(vii) |
37 |
Processes are in place to assure the management of outsourced activities. |
Figure 2 and Table 2 address the 10 most frequently cited requirements in Chapter 4. The most frequent citation addresses contemporaneous documentation of data and information, and the requirement that these actions are traceable. Document management is also among the most frequent citations.
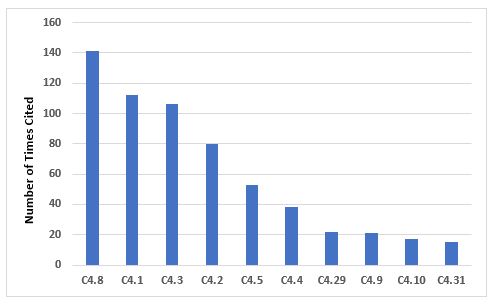
Figure 2: Top 10 deficiencies citing Chapter 4
Table 2: Top 10 Deficiencies Citing Chapter 4
|
Paragraph |
# |
Short Text |
|
4.8 |
141 |
Records should be made or completed at the time each action is taken and in such a way that all significant activities concerning the manufacture of medicinal products are traceable. |
|
4.1 |
112 |
All types of document should be defined and adhered to. The requirements apply equally to all forms of document media types. Complex systems need to be understood, well documented, validated, and adequate controls should be in place… |
|
4.3 |
106 |
Documents containing instructions should be approved, signed, and dated by appropriate and authorised persons. Documents should have unambiguous contents and be uniquely identifiable. The effective date should be defined. |
|
4.2 |
80 |
Documents should be designed, prepared, reviewed, and distributed with care… |
|
4.5 |
53 |
Documents within the Quality Management System should be regularly reviewed and kept up to date. |
|
4.4 |
38 |
Documents containing instructions should be laid out in an orderly fashion and be easy to check… |
|
4.29 |
22 |
There should be written policies, procedures, protocols, reports, and the associated records of actions taken or conclusions reached, where appropriate, for the following examples: … |
|
4.9 |
21 |
Any alteration made to the entry on a document should be signed and dated; the alteration should permit the reading of the original information. Where appropriate, the reason for the alteration should be recorded. |
|
4.10 |
17 |
It should be clearly defined which record is related to each manufacturing activity and where this record is located. Secure controls must be in place to ensure the integrity of the record throughout the retention period and validated where appropriate. |
|
4.31 |
15 |
Logbooks should be kept for major or critical analytical testing, production equipment, and areas where product has been processed… |
Figure 3 and Table 3 identify the 10 most frequent deficiencies that cite Chapter 5. Many of the requirements are cited with a similar frequency, thus no single requirement stands out as with some other areas.
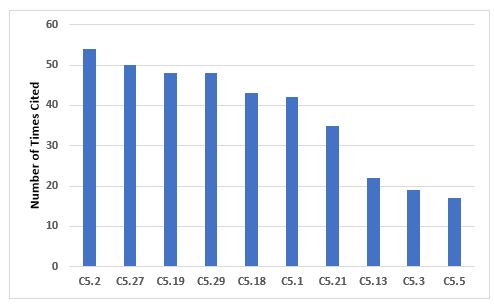
Figure 3: Top 10 deficiencies citing Chapter 5
Table 3: Top 10 Deficiencies Citing Chapter 5
|
Paragraph |
# |
Short Text |
|
5.2 |
54 |
All handling of materials and products, such as receipt and quarantine, sampling, storage, labelling, dispensing, processing, packaging, and distribution should be done in accordance with written procedures or instructions and, where necessary, recorded. |
|
5.27 |
50 |
The selection, qualification, approval, and maintenance of suppliers of starting materials, together with their purchase and acceptance, should be documented as part of the pharmaceutical quality system… |
|
5.19 |
48 |
Cross-contamination should be prevented by attention to design of the premises and equipment as described in Chapter 3… |
|
5.29 |
48 |
For the approval and maintenance of suppliers of active substances and excipients, the following is required: ... |
|
5.18 |
43 |
Contamination of a starting material or of a product by another material or product should be prevented… |
|
5.1 |
42 |
Production should be performed and supervised by competent people. |
|
5.21 |
35 |
The outcome of the Quality Risk Management process should be the basis for determining the extent of technical and organisational measures required to control risks for cross-contamination. These could include, but are not limited to, the following: … |
|
5.13 |
22 |
Labels applied to containers, equipment, or premises should be clear, unambiguous, and in the company’s agreed format… |
|
5.3 |
19 |
All incoming materials should be checked to ensure that the consignment corresponds to the order… |
|
5.5 |
17 |
Incoming materials and finished products should be physically or administratively quarantined immediately after receipt or processing, until they have been released for use or distribution. |
Annex 15, Qualification and Validation
Figure 4 and Table 4 show the 10 most frequent requirements cited in Annex 15. The most frequently cited requirement includes the need to identify and investigate deviations, including failures to meet acceptance criteria, once again emphasizing the importance of this area, which is also identified as the most frequent citation of Chapter 1 requirements.
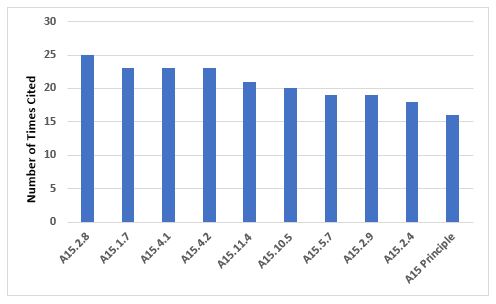
Figure 4: Top 10 deficiencies citing Annex 15
Table 4: Top 10 Deficiencies Citing Annex 15
|
Paragraph |
# |
Short Text |
|
15.2.8 |
25 |
Results which fail to meet the predefined acceptance criteria should be recorded as a deviation and be fully investigated according to local procedures. Any implications for the validation should be discussed in the report. |
|
15.1.7 |
23 |
A quality risk management approach should be used for qualification and validation activities… |
|
15.4.1 |
23 |
A quality risk management approach should be used for qualification and validation activities. |
|
15.4.2 |
23 |
Where re-qualification is necessary and performed at a specific time period, the period should be justified and the criteria for evaluation defined… |
|
15.11.4 |
21 |
Quality risk management should be used to evaluate planned changes to determine the potential impact on product quality, pharmaceutical quality systems, documentation, validation, regulatory status, calibration, maintenance, and on any other system to avoid unintended consequences and to plan for any necessary process validation, verification, or requalification efforts. |
|
15.10.5 |
20 |
For all cleaning processes an assessment should be performed to determine the variable factors which influence cleaning effectiveness and performance, e.g., operators, the level of detail in procedures such as rinsing times etc. … |
|
15.5.7 |
19 |
Process validation should establish whether all quality attributes and process parameters, which are considered important for ensuring the validated state and acceptable product quality, can be consistently met by the process. |
|
15.2.9 |
19 |
The review and conclusions of the validation should be reported and the results obtained summarised against the acceptance criteria… |
|
15.2.4 |
18 |
Validation protocols should be prepared which defines the critical systems, attributes and parameters and the associated acceptance criteria. |
|
Principle |
16 |
This Annex describes the principles of qualification and validation which are applicable to the facilities, equipment, utilities, and processes used for the manufacture of medicinal products and may also be used as supplementary optional guidance for active substances without introduction of additional requirements to EudraLex, Volume 4, Part II… |
Chapter 3, Premises and Equipment
Figure 5 and Table 5 identify the 10 most frequent deficiencies in Chapter 3. The three most frequently cited requirements include the need to have adequate storage areas with appropriate environmental controls, secure storage of highly active materials, and the need to calibrate and maintain weighing and measuring equipment.
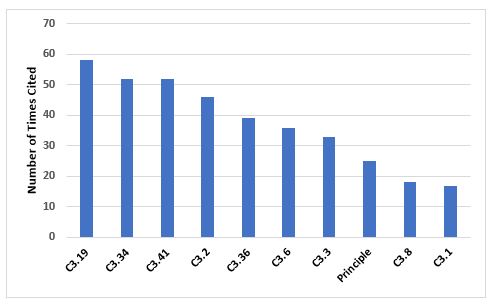
Figure 5: Top 10 deficiencies citing Chapter 3
Table 5: Top 10 Deficiencies Citing Chapter 3
|
Paragraph |
# |
Short Text |
|
3.19 |
58 |
Storage areas should be designed or adapted to ensure good storage conditions. In particular, they should be clean and dry and maintained within acceptable temperature limits… |
|
3.34 |
52 |
Highly active materials or products should be stored in safe and secure areas. |
|
3.41 |
52 |
Measuring, weighing, recording and control equipment should be calibrated and checked at defined intervals by appropriate methods… |
|
3.2 |
46 |
Premises should be carefully maintained, ensuring that repair and maintenance operations do not present any hazard to the quality of products… |
|
3.36 |
39 |
Manufacturing equipment should be designed so that it can be easily and thoroughly cleaned… |
|
3.6 |
36 |
Cross-contamination should be prevented for all products by appropriate design and operation of manufacturing facilities… |
|
3.3 |
33 |
Lighting, temperature, humidity and ventilation should be appropriate and such that they do not adversely affect, directly or indirectly, either the medicinal products during their manufacture and storage, or the accurate functioning of equipment. |
|
Principle |
25 |
Premises and equipment must be located, designed, constructed, adapted and maintained to suit the operations to be carried out... |
|
3.8 |
18 |
The adequacy of the working and in-process storage space should permit the orderly and logical positioning of equipment and materials so as to minimise the risk of confusion between different medicinal products or their components, to avoid cross contamination and to minimise the risk of omission or wrong application of any of the manufacturing or control steps. |
|
3.1 |
17 |
Premises should be situated in an environment which, when considered together with measures to protect the manufacture, presents minimal risk of causing contamination of materials or products. |
Annex 1, Manufacture of Sterile Medicinal Products
Figure 6 and Table 6 identify the 14 most frequent requirements cited in Annex 1. The most frequent citation addresses precautions to avoid contamination and the second addresses environmental monitoring in the areas where aseptic operations are performed. The third most frequent requirement cited is associated with setting appropriate alert and action limits for the results of environmental monitoring. Five requirements are tied for 10th place, so they are all included.
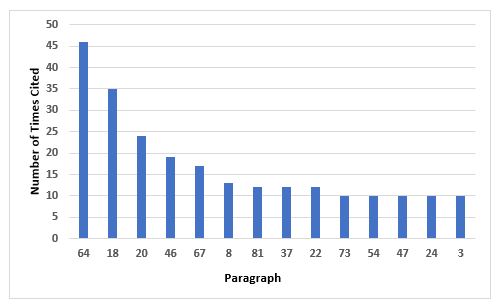
Figure 6: Top 10 deficiencies citing Annex 1
Table 6: Top 10 Deficiencies Citing Annex 1
|
Paragraph |
# |
Short Text |
|
64 |
46 |
Precautions to minimize contamination should be taken during all processing stages including the stages before sterilisation. |
|
18 |
35 |
Where aseptic operations are performed monitoring should be frequent using methods such as settle plates, volumetric air and surface sampling (e.g. swabs and contact plates). Sampling methods used in operation should not interfere with zone protection… |
|
20 |
24 |
Appropriate alert and action limits should be set for the results of particulate and microbiological monitoring. If these limits are exceeded operating procedures should prescribe corrective action. |
|
46 |
19 |
In clean areas, all exposed surfaces should be smooth, impervious and unbroken in order to minimize the shedding or accumulation of particles or micro-organisms and to permit the repeated application of cleaning agents, and disinfectants where used. |
|
67 |
17 |
The process simulation test should imitate as closely as possible the routine aseptic manufacturing process and include all the critical subsequent manufacturing steps… |
|
8 |
13 |
Clean rooms and clean air devices should be routinely monitored in operation and the monitoring locations based on a formal risk analysis study and the results obtained during the classification of rooms and/or clean air devices. |
|
81 |
12 |
Components, containers, equipment and any other article required in a clean area where aseptic work takes place should be sterilised and passed into the area through double-ended sterilisers sealed into the wall, or by a procedure which achieves the same objective of not introducing contamination… |
|
37 |
12 |
All personnel (including those concerned with cleaning and maintenance) employed in such areas should receive regular training in disciplines relevant to the correct manufacture of sterile products… |
|
22 |
12 |
The transfer of materials into and out of the unit is one of the greatest potential sources of contamination… |
|
73 |
10 |
Activities in clean areas and especially when aseptic operations are in progress should be kept to a minimum and movement of personnel should be controlled and methodical, to avoid excessive shedding of particles and organisms due to over-vigorous activity… |
|
54 |
10 |
It should be demonstrated that air-flow patterns do not present a contamination risk, e.g. care should be taken to ensure that air flows do not distribute particles from a particle generating person, operation or machine to a zone of higher product risk. |
|
47 |
10 |
To reduce accumulation of dust and to facilitate cleaning there should be no uncleanable recesses and a minimum of projecting ledges, shelves, cupboards and equipment… |
|
24 |
10 |
Isolators should be introduced only after appropriate validation… |
|
3 |
10 |
Clean areas for the manufacture of sterile products are classified according to the required characteristics of the environment… |
Figure 7 and Table 7 identify the most frequently cited requirements in Chapter 6. The two most frequently cited requirements are the need to adequately control laboratory components and reagents and the need to trend data and investigate OOT (out of trend) and OOS (out of specification) events. Again, we find the importance of investigating such events similar to those identified in deficiencies cited in Chapter 5 and Chapter 1.
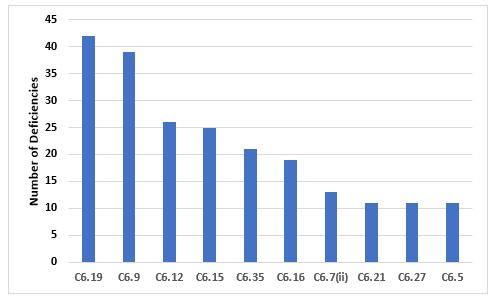
Figure 7: Top 10 deficiencies citing Chapter 6
Table 7: Top 10 Deficiencies Citing Chapter 6
|
Paragraph |
# |
Short Text |
|
6.19 |
42 |
Special attention should be given to the quality of laboratory reagents, solutions, glassware, reference standards, and culture media… |
|
6.9 |
39 |
Some kinds of data (e.g., tests results, yields, environmental controls) should be recorded in a manner permitting trend evaluation. Any out of trend or out of specification data should be addressed and subject to investigation. |
|
6.12 |
26 |
Samples should be representative of the batch of materials or products from which they are taken… |
|
6.15 |
25 |
Testing methods should be validated… |
|
6.35 |
21 |
Out of specification or significant atypical trends should be investigated. Any confirmed out of specification result, or significant negative trend, affecting product batches released on the market should be reported to the relevant competent authorities… |
|
6.16 |
19 |
The results obtained should be recorded. Results of parameters identified as quality attribute or as critical should be trended and checked to make sure that they are consistent with each other. Any calculations should be critically examined. |
|
6.7(ii) |
13 |
Procedures describing sampling, testing, records (including test worksheets and/or laboratory notebooks), recording, and verifying; |
|
6.21 |
11 |
Laboratory reagents, solutions, reference standards, and culture media should be marked with the preparation and opening date and the signature of the person who prepared them… |
|
6.27 |
11 |
The purpose of the ongoing stability programme is to monitor the product over its shelf life and to determine that the product remains, and can be expected to remain, within specifications under the labelled storage conditions. |
|
6.5 |
11 |
Control laboratory premises and equipment should meet the general and specific requirements for Quality Control areas given in Chapter 3… |
Chapter 8, Complaints and Product Recall
Figure 8 and Table 8 identify the top 10 deficiencies that cite Chapter 8. By far the most frequent citation addresses the need to periodically challenge and determine the effectiveness of the recall process. Generally, this exercise is expected to be conducted annually. The next two most frequent deficiencies address the need to implement appropriate corrective and preventive actions in response to quality defects and to report qualify defects to the health authorities when this may result in a product recall.
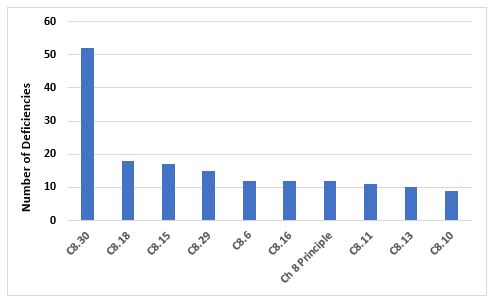
Figure 8: Top 10 deficiencies citing Chapter 8
Table 8: Top 10 Deficiencies Citing Chapter 8
|
Paragraph |
# |
Short Text |
|
8.30 |
52 |
The effectiveness of the arrangements in place for recalls should be periodically evaluated to confirm that they remain robust and fit for use… |
|
8.18 |
18 |
Appropriate CAPAs should be identified and taken in response to a quality defect. The effectiveness of such actions should be monitored and assessed. |
|
8.15 |
17 |
Quality defects should be reported in a timely manner by the manufacturer to the marketing authorisation holder/sponsor and all concerned Competent Authorities in cases where the quality defect may result in the recall of the product or in an abnormal restriction in the supply of the product. |
|
8.29 |
15 |
The progress of the recall process should be recorded until closure and a final report issued, including a reconciliation between the delivered and recovered quantities of the concerned products/batches. |
|
8.6 |
12 |
Special attention should be given to establishing whether a complaint or suspected quality defect relates to falsification. |
|
8.16 |
12 |
An appropriate level of root cause analysis work should be applied during the investigation of quality defects… |
|
Principle |
12 |
In order to protect public and animal health, a system and appropriate procedures should be in place to record, assess, investigate, and review complaints, including potential quality defects, and if necessary, to effectively and promptly recall medicinal products for human or veterinary use and investigational medicinal products from the distribution network… |
|
8.11 |
11 |
If a quality defect is discovered or suspected in a batch, consideration should be given to checking other batches and, in some cases, other products, in order to determine whether they are also affected… |
|
8.13 |
10 |
The decisions that are made during and following quality defect investigations should reflect the level of risk that is presented by the quality defect as well as the seriousness of any non-compliance with respect to the requirements of the marketing authorisation/product specification file or GMP… |
|
8.10 |
9 |
When a quality defect investigation is initiated, procedures should be in place to address at least the following: … |
Chapter 7, Outsourced Activities
Figure 9 and Table 9 show the deficiencies that cite Chapter 7. Among the two most frequent citations is the need for a Quality Agreement and the content of that agreement with regard to the outsourced activity.
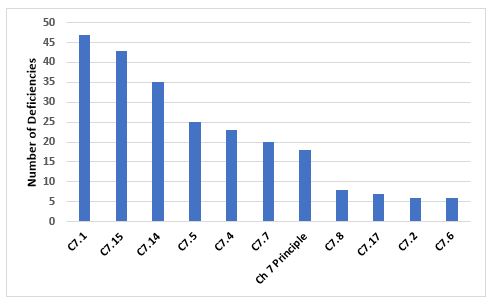
Figure 9: Top 10 deficiencies citing Chapter 7
Table 9: Top 10 deficiencies citing Chapter 7
|
Paragraph |
# |
Short Text |
|
7.1 |
47 |
There should be a written Contract covering the outsourced activities, the products or operations to which they are related, and any technical arrangements made in connection with it. |
|
7.15 |
43 |
The Contract should describe clearly who undertakes each step of the outsourced activity, e.g., knowledge management, technology transfer, supply chain, subcontracting, quality and purchasing of materials, testing and releasing materials, undertaking production, and quality controls (including in-process controls, sampling and analysis). |
|
7.14 |
35 |
A Contract should be drawn up between the Contract Giver and the Contract Acceptor which specifies their respective responsibilities and communication processes relating to the outsourced activities. |
|
7.5 |
25 |
Prior to outsourcing activities, the Contract Giver is responsible for assessing the legality, suitability and the competence of the Contract Acceptor to carry out successfully the outsourced activities… |
|
7.4 |
23 |
The pharmaceutical quality system of the Contract Giver should include the control and review of any outsourced activities… |
|
7.7 |
20 |
The Contract Giver should monitor and review the performance of the Contract Acceptor and the identification and implementation of any needed improvement. |
|
Principle |
18 |
Any activity covered by the GMP Guide that is outsourced should be appropriately defined, agreed and controlled in order to avoid misunderstandings which could result in a product or operation of unsatisfactory quality… |
|
7.8 |
8 |
The Contract Giver should be responsible for reviewing and assessing the records and the results related to the outsourced activities… |
|
7.17 |
7 |
The Contract should permit the Contract Giver to audit outsourced activities, performed by the Contract Acceptor or his mutually agreed subcontractors |
|
7.2 |
6 |
All arrangements for the outsourced activities including any proposed changes in technical or other arrangements should be in accordance with regulations in force, and the Marketing Authorisation for the product concerned, where applicable. |
Annex 11, Computerized Systems
The 10 most frequent inspection deficiencies that identify requirements in Annex 11 are shown in Figure 10 and Table 10. The first most frequent deficiency addresses the application of risk management throughout the life cycle of computerized systems. The second most frequent deficiency addresses the failure to ensure integrity of that data and that it is routinely backed up.
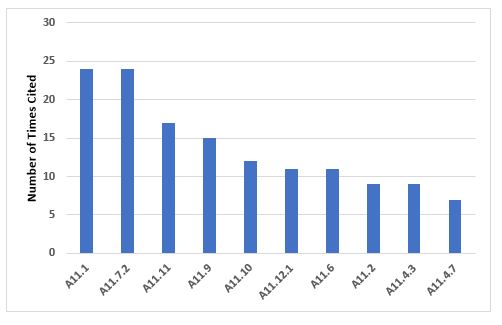
Figure 10: Top 10 deficiencies citing Annex 11
Table 10: Top 10 Deficiencies Citing Annex 11
|
Paragraph |
# |
Short Text |
|
11.1 |
24 |
Risk management should be applied throughout the life cycle of the computerized system taking into account patient safety, data integrity and product quality… |
|
11.7.2 |
24 |
Regular backups of all relevant data should be done. Integrity and accuracy of backup data and the ability to restore the data should be checked during validation and monitored periodically. |
|
11.11 |
17 |
Computerized systems should be periodically evaluated to confirm that they remain in a valid state and are compliant with GMP. Such evaluations should include, where appropriate, the current range of functionality, deviation records, incidents, problems, upgrade history, performance, reliability, security and validation status reports… |
|
11.9 |
15 |
Consideration should be given, based on a risk assessment, to building into the system the creation of a record of all GMP-relevant changes and deletions (a system generated "audit trail") … |
|
A1.10 |
12 |
Any changes to a computerized system including system configurations should only be made in a controlled manner in accordance with a defined procedure. |
|
11.12.1 |
11 |
Physical and/or logical controls should be in place to restrict access to computerized system to authorised persons… |
|
11.6 |
11 |
For critical data entered manually, there should be an additional check on the accuracy of the data… |
|
11.2 |
9 |
There should be close cooperation between all relevant personnel such as Process Owner, System Owner, Qualified Persons, and IT… |
|
11.4.3 |
9 |
An up to date listing of all relevant systems and their GMP functionality (inventory) should be available. |
|
11.4.7 |
7 |
Evidence of appropriate test methods and test scenarios should be demonstrated. Particularly, system (process) parameter limits, data limits and error handling should be considered… |
Conclusion
This two-part article identified the MHRA drug product GMP deficiencies for 2018, based on a 6200+ row spreadsheet issued by the agency. Overall conclusions include:
- The number of MHRA inspections decreased substantially in 2018 and is lower than it was in 2015.
- During the three years addressed in Part 1, 75 to 80 percent of inspections occurred the UK, and 20 to 25 percent occurred in other countries.
- While some changes have occurred over that time period, Pharmaceutical Quality System (Chapter 1) has the largest number of deficiencies in each of the three years.
- Critical deficiencies account for only 2 percent of the total, major deficiencies for approximately 40 percent, and “other” deficiencies make up the remaining almost 60 percent.
- Critical deficiencies cluster in Chapter 1 and Annex 1
- Chapter 1 again leads the way for major deficiencies, and Annex 15, Annex 1, and Chapter 5 are a distant second through fourth.
When considering the top 10 paragraphs among the top 10 chapters and annexes cited, the issue of investigations and root cause analysis appears frequently. This is similar to the FDA’s focus on deviations and investigations. The lack of granularity in the FDA regulations makes comparison to the MHRA deficiencies a challenge, but at a high level they are similar, with a focus on the importance of quality systems and the quality unit, and investigations and assignment of root cause. Firms would be well served to evaluate the MHRA areas of focus and deficiencies to assist in inspection preparation as well as Qualified Person (QP) audits that European based firms may conduct, particularly for business partners and CMOs.
We appreciate MHRA’s publication of these data and encourage them to continue — and perhaps publish their documentation (GDP) and clinical (GCP) deficiencies in a similar format.
Acknowledgements:
I want to acknowledge the kind assistance provided by Eileen Counihan, Zachary Unger, and Clare Nolan in helping me learn and negotiate the pivot tables that made this article possible.
About The Author:
 Barbara Unger formed Unger Consulting, Inc. to provide GMP auditing and regulatory intelligence services to the pharmaceutical industry, including general GMP auditing and auditing and remediation in the area of data management and data integrity. Her auditing experience includes leadership of the Amgen corporate GMP audit group for APIs and quality systems. She also developed, implemented, and maintained the GMP regulatory intelligence program for eight years at Amgen. This included surveillance, analysis, and communication of GMP related legislation, regulations, guidance, and industry compliance enforcement trends. Unger was the first chairperson of the Rx-360 Monitoring and Reporting work group that summarized and published relevant GMP and supply chain related laws, regulations, and guidance. She co-led the Rx-360 Data Integrity Working Group from 2017–2019. You can contact her at bwunger123@gmail.com.
Barbara Unger formed Unger Consulting, Inc. to provide GMP auditing and regulatory intelligence services to the pharmaceutical industry, including general GMP auditing and auditing and remediation in the area of data management and data integrity. Her auditing experience includes leadership of the Amgen corporate GMP audit group for APIs and quality systems. She also developed, implemented, and maintained the GMP regulatory intelligence program for eight years at Amgen. This included surveillance, analysis, and communication of GMP related legislation, regulations, guidance, and industry compliance enforcement trends. Unger was the first chairperson of the Rx-360 Monitoring and Reporting work group that summarized and published relevant GMP and supply chain related laws, regulations, and guidance. She co-led the Rx-360 Data Integrity Working Group from 2017–2019. You can contact her at bwunger123@gmail.com.