
The Hidden Engineering Behind Successful Upstream Bioprocessing
By Smiriti Gupta, NIRAS

The global healthcare landscape is undergoing a profound transformation. The rapid rise of biologics, biosimilars, and personalized medicine has positioned biopharmaceuticals at the leading edge of this change. These therapies are more targeted, more complex, and more sensitive than traditional small molecule drugs.1
Unlike chemical pharmaceuticals, which are synthesized through organic chemistry, biopharmaceuticals are derived from living systems such as bacteria, yeast, or mammalian cells. They include monoclonal antibodies, recombinant proteins, vaccines, gene therapies, and mRNA‑based products. Because the factory is biological, manufacturing is inseparable from biology, engineering, contamination control, and quality science.
This article consolidates insights from knowledge-sharing sessions, which I led through my work at NIRAS.
Chemical Pharmaceuticals Vs. Biopharmaceuticals: A Fundamental Shift
At the heart of any medicine lies the active pharmaceutical ingredient (API). The distinction between chemical and biological APIs defines how products are developed, manufactured, and controlled.
Chemical pharmaceuticals (small molecules) are:
- synthesized through defined chemical reactions,
- mostly low molecular weight and structurally simple, and
- generally stable and reproducible.
- Examples: ibuprofen, paracetamol, statins.
These drugs typically work by inhibiting or activating biochemical pathways through receptor or enzyme binding.
Biopharmaceuticals (large molecules) are:
- produced by living cells (microbial, yeast, or mammalian),
- high molecular weight and structurally complex, and
- sensitive to temperature, shear, and contamination.
- Examples: insulin, monoclonal antibodies, vaccines, gene and cell therapies.
Rather than being synthesized, biologics are expressed by living cells. Their therapeutic action often mimics or enhances natural biological processes, offering higher specificity and, in many cases, fewer side effects.
Why This Distinction Matters
Because biologics depend on living systems, their manufacturing demands:
- highly controlled, sterile environments,
- sophisticated cell culture and bioprocess engineering,
- advanced purification technologies, and
- rigorous cleaning, sterilization, and regulatory compliance.
This complexity is what makes biopharma manufacturing both challenging and critically important.
The Biopharmaceutical Manufacturing Life Cycle: An Integrated View
The journey from gene to patient unfolds through a tightly controlled and interconnected sequence of steps. Broadly, the life cycle includes:
- upstream processing (USP) – cell cultivation and product generation,
- downstream processing (DSP) – recovery and purification of the product, and
- drug product manufacturing (DP) – formulation, aseptic filling, and packaging.
Supporting these core stages are essential enabling systems such as cleaning‑in‑place (CIP), sterilization‑in‑place (SIP), and biowaste deactivation, which ensure patient safety, environmental protection, and regulatory compliance.
Cleanroom Strategy: Aligning Classification To Risk
Cleanroom strategy is not an afterthought in biomanufacturing. It is a design choice that directly shapes contamination risk, operability, and cost. Biotech operations typically run at moderate temperature and pressure, but they demand tightly controlled particulate and microbial conditions across suites, corridors, and aseptic zones.
- Class D (entry/change areas): commonly applied for change rooms and entry rooms into manufacturing suites.
- Class C (controlled support areas): often used for corridors and clean‑utility areas.
- Class B (background to aseptic processing): used around critical operations and tightly controlled processing spaces.
- Class A (critical aseptic zone): applies to highest sterility areas such as filling; typically surrounded by Class B background.
Automation As The Backbone Of GMP Execution
As facilities scale, automation becomes the lifeblood connecting GMP compliance and efficient operations. A typical biotech plant uses layered automation across utilities, process units, and facility systems so recipes, alarms, data integrity, and batch reporting remain consistent end‑to‑end.
- Packaged units (smart skids) often run their own PLC control and integrate to a DCS for central oversight.
- Facility automation includes Heating, ventilation, and air conditioning (HVAC) and building systems such as Building Management System (BMS)/ Environmental Monitoring system (EMS), plus interfaces to biowaste handling (kill tank/ Effluent Treatment Plant (ETP)).
- Utility automation covers clean utilities (purified water/WFI, clean steam, process gases/air) and black utilities (steam boiler, chiller, compressors, cooling tower, raw water).
- Plant/process automation spans drug substance (upstream + downstream) and drug product (formulation, filling, and packaging).
The Approval Pathway: Why Development Shapes Manufacturing
Manufacturing decisions are also shaped by the development and approval pathway. From discovery to post‑marketing surveillance, clinical phases progressively expand from small safety studies to large, long‑term monitoring — while manufacturing controls, validation, and quality systems mature in parallel.
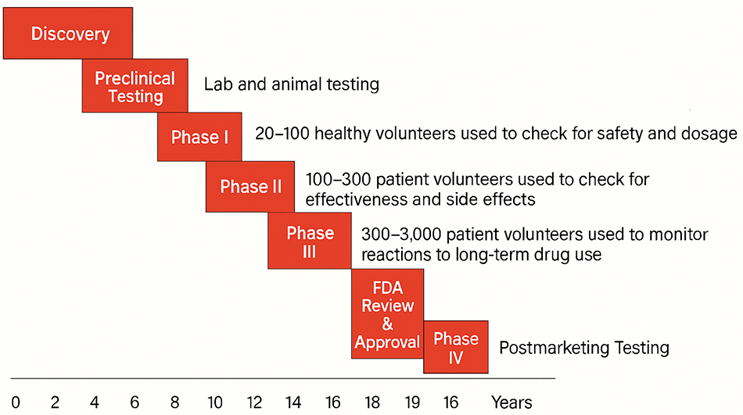
FDA Approval Pathway2
Biopharma Process Characteristics: Why Operations Look Different
Compared with chemical pharma, biotechnological manufacturing has a distinct operating fingerprint: it is predominantly aqueous, highly contamination‑sensitive, and economically shaped by smaller annual volumes and a much higher cost‑per‑kilogram of active substance. These characteristics explain why biopharma plants are typically more capital‑intensive and why contamination control and biosafety sit at the center of design.
- They are manufactured under stringent oversight (e.g., FDA/WHO‑aligned expectations).2
- Their safety profile is skewed toward biohazards (rather than chemical hazards).1
- Higher capital intensity: New biologics capacity frequently requires hundreds of millions to ~$2 billion and long lead times, materially above typical chemical API plants.3
- Their active substances are very high value. Treatment‑level pricing for mAbs often normalizes to $2,000/g on the low end, with much higher per‑gram equivalents in some indications.4
- They have smaller typical production volumes. Biologics are high‑value, low‑tonnage products relative to small molecules, due to potency and complex processing.
- Cleanroom‑based operations. Aseptic steps (e.g., filling, critical transfers) require classified cleanrooms and environmental controls per FDA CGMP guidance.5
- They rely on predominantly aqueous processing (minimal solvent use).

The Manufacturing Floor, Simplified.
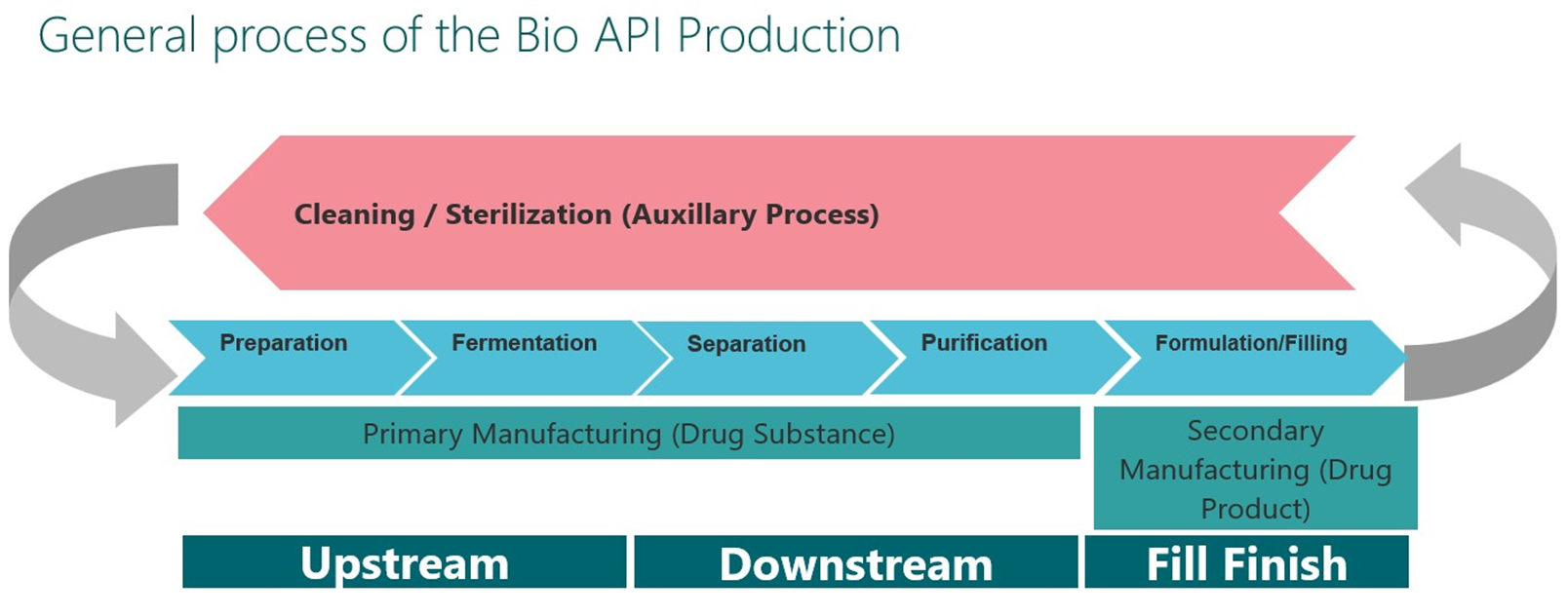
Upstream processing: creating the living factory
Upstream processing is the biological heart of biopharmaceutical manufacturing. Its purpose is to generate the desired protein or biological product by cultivating living cells under tightly controlled conditions. Operational robustness downstream is fundamentally dependent on the quality and consistency established here.
Cell line, media, and inoculum preparation
Every biopharmaceutical product originates from a well‑characterized, cryopreserved cell line stored in a master and working cell bank. Manufacturing begins with:
- Thawing the selected cell strain under controlled conditions.
- Transfer into an inoculation chamber/laminar air flow unit (LAFU) to maintain sterility.
- Initial expansion in shake flasks, typically performed manually.
In parallel, media preparation takes place in the manufacturing area. Media consists of precisely defined nutrients, salts, and growth factors dissolved in sterile water. Depending on process design, media is:
- filter‑sterilized (for heat‑sensitive components) or
- steam‑sterilized (for robust formulations).
- smaller quantities can also be autoclaved (very common for feeds and complimentary additions)
Only once both inoculum and sterile media are ready does the production process begin.
Seed vs. production fermentation: controlled scale‑up
Fermentation rarely occurs in a single step. Instead, it follows a multi‑stage scale‑up strategy (also called as seed-train in some places):
Seed Fermentation
- It is conducted in multiple progressively larger seed fermenters.
- The objective is cell mass amplification, not product formation.
- Each seed fermenter acts as the inoculum for the next stage. This ensures cells are physiologically adapted before large‑scale production.
Production Regime
- The final stage is large‑volume fermenter/bioreactor.
- The primary objective is product expression rather than growth alone.
- Process parameters are tightly controlled to favor product synthesis. This staged approach minimizes biological stress and ensures reproducibility at production scale.
During fermentation, cells are cultivated under continuously monitored conditions:
- pH
- Dissolved oxygen
- Temperature
- Agitation and aeration
- Nutrient and feed rates
- Redox Potential (Secondary Parameter)
Note: Its effects are managed indirectly through established controls such as dissolved oxygen, aeration, agitation, and feeding.
Cell growth follows four classical phases:
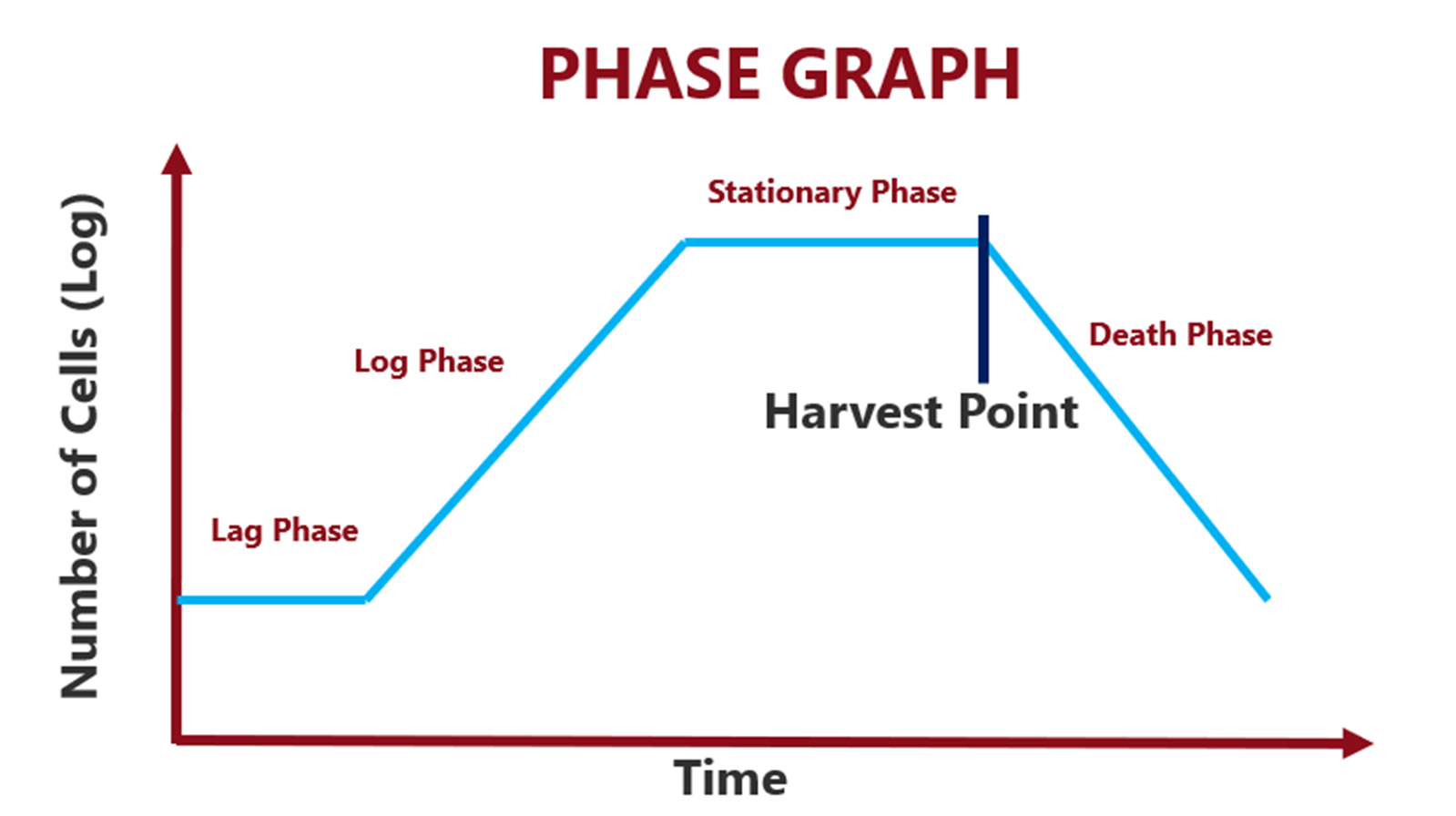
- Lag phase – Cellular adaptation with no increase in cell count.
- Exponential (log) phase – Rapid cell division and biomass increase.
- Stationary phase – Growth rate equals cell death rate.
- Death phase – Nutrient depletion and toxic metabolite accumulation.
Harvesting is deliberately performed just before the death phase, maximizing yield while preserving product integrity.
Modern fermenters are equipped with sensors, control valves, and automation systems that allow real‑time adjustments and consistent batch‑to‑batch performance.
What To Expect In Part 2
The next article in this series takes you beyond the fermenter into the mechanical and separation-focused world of downstream processing. It details harvest operations, clarification methods, such as centrifugation and depth filtration, and critical unit operations like tangential flow filtration (ultrafiltration/diafiltration) used to prepare material for purification. It then walks through chromatography strategies, viral clearance (where applicable), and finally transitions into drug substance handling, formulation, sterile filtration, aseptic filling, and lyophilization. Part 2 completes the journey from living cells to a GMP-compliant, patient-ready biologic.
References:
- Biological Product Definitions, FDA, https://www.fda.gov/files/drugs/published/Biological-Product-Definitions.pdf
- FDA. The Drug Development Process. U.S. Food & Drug Administration, https://www.fda.gov/patients/learn-about-drug-and-device-approvals/drug-development-process
- Biopharmaceutical Manufacturing, PhRMA, https://phrma.org/policy-issues/research-development/biopharmaceutical-manufacturing
- Chun Chen, et al., "Cost and supply considerations for antibody therapeutics", mAbs, doi.org/10.1080/19420862.2025.2451789
- Sterile Drug Products Produced by Aseptic Processing — Current Good Manufacturing Practice , FDA, https://www.fda.gov/regulatory-information/search-fda-guidance-documents/sterile-drug-products-produced-aseptic-processing-current-good-manufacturing-practice
About The Author:
 Smiriti Gupta is a senior consultant with NIRAS International Consulting in Denmark where she focuses on process automation and optimization; commissioning, qualification, and validation; and life cycle management. Previously she worked at WuXi Biologics as a computerized system validation engineer, at Siemens as a technical support specialist, and for DD Enterprises as a senior bioprocess and automation manager. Her career began as an operator on the biopharma manufacturing floor, giving her first‑hand, shop‑floor perspective that directly informs the insights shared in this article. She has a Bachelor of Engineering degree from the Madhav Institute of Technology and Science.
Smiriti Gupta is a senior consultant with NIRAS International Consulting in Denmark where she focuses on process automation and optimization; commissioning, qualification, and validation; and life cycle management. Previously she worked at WuXi Biologics as a computerized system validation engineer, at Siemens as a technical support specialist, and for DD Enterprises as a senior bioprocess and automation manager. Her career began as an operator on the biopharma manufacturing floor, giving her first‑hand, shop‑floor perspective that directly informs the insights shared in this article. She has a Bachelor of Engineering degree from the Madhav Institute of Technology and Science.