
The Essential Components Of A Sterility Assurance Program
By Crystal M. Booth, PSC Biotech

Sterility assurance is a level of confidence that a particular product or unit that is purported to be sterile is sterile. Sterility cannot be demonstrated without the destruction of every unit of product produced. Therefore, sterility assurance is achieved through multiple practices and procedures. This article discusses the different variables of contamination control that help to increase confidence in sterility assurance.
USP <1211> is a general information chapter on sterility assurance. The chapter states that “an item is deemed sterile only when it contains no viable microorganisms.”1 It is well known in the industry that the sterility testing model described in USP <71> Sterility Tests has limitations. The test only indicates that the subset of articles from a lot that are tested are sterile. The test is destructive in that every unit that is tested is either consumed or no longer sterile after the test is performed. To help ensure consumer safety, additional measures must be put into place to add assurance that the entire batch or lot of products manufactured is sterile. This is accomplished using a sterility assurance program.
Many companies have sterility assurance programs in place but may not have their processes labeled as a sterility assurance program. Figure 1 from USP <1211> shows several factors that influence sterility assurance. The factors listed in Figure 1 should be considered for their impact on the sterility of the final product.
Figure 1: Influences on Sterile Products.
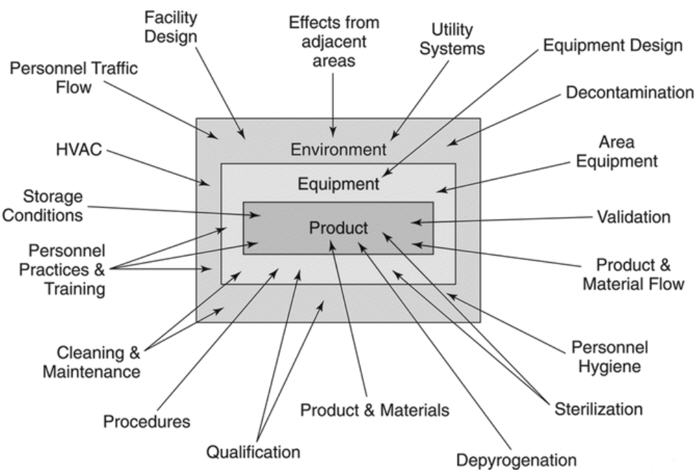
The first step in developing a sterility assurance program is to list each step in the process, beginning at the point of use and ending in sterile storage.2 Each step should be evaluated for ways to prevent contamination in the manufacturing process or environments.
A sterility assurance program should be fit for purpose for the product or device that is being manufactured. A holistic sterility assurance program for an aseptic manufactured product could include the following components.
Personnel
Personnel must be properly trained, educated, and/or supervised to be involved with aseptic processing. The training records must be maintained. Training concepts should include the importance of proper aseptic technique and cleanroom behaviors. It must be recognized that humans are the primary source of contamination in the cleanroom environment. Retraining and qualification of personnel should be done on a routine basis to keep personnel sensitized to the importance of aseptic technique.3
Personal Hygiene and Sanitation Practices
There must be procedures and training that govern personnel hygiene, sanitation, aseptic technique, aseptic behavior in the cleanrooms, and aseptic gowning practices. Personnel must adhere to sanitation and health precautions designed to avoid contamination of the test, environment, and/or product. Personnel must also adhere to gowning and personal protective equipment procedures. If an employee is feeling ill, they must inform their supervisor of any health or medical condition that may have an adverse effect on a test, product, or environment. Personnel must also be monitored for microbial growth and undergo gowning qualification training to ensure aseptic status of the manufacturing or testing environment.
Personnel Flow
There must be procedures and practices regarding personnel flow. Personnel must follow established entry and exit routes to prevent cross contamination. The routes should include different levels of gowning for each grade of the cleanroom environment. These routes must also be established in standard operating procedures (SOPs) and understood by personnel.
Procedures
All procedures for aseptic processing and subsequent quality control testing must be written in accordance with good manufacturing practices (GMPs). Procedures must remain up to date, accurate, and revised when warranted.
Product and Material Flow
Like the procedures and practices regarding personnel flow, there must be procedures and practices for product and material flow to prevent cross contamination. The routes should include levels or methods of sanitization of products, materials, and/or waste as they enter or exit the cleanroom areas. These routes must also be established in standard operating procedures and understood by personnel.
Equipment
The use and preparation of equipment for aseptic processing must be documented in SOPs. It must be designed appropriately for the intended use and housed in a manner to prevent cross contamination. Equipment used in the generation, measurement, or assessment of data and equipment used for facility environmental control must be of the specified design and capacity to function according to GMPs. The equipment must be suitably located for operation, inspection, cleaning, and maintenance. It must be inspected, cleaned, and maintained. Equipment used for the generation, measurement, or assessment of data must be tested, calibrated, standardized, and/or sterilized.
Sterilization
The use and sterilization of equipment, components, or other materials for aseptic processing must be governed in SOPs. This could include purchasing items that are ready to use or preparing the items for use in-house.
Depyrogenation
Likewise, the use and depyrogenation of equipment, components, or other materials for aseptic processing must be governed in SOPs. This could include purchasing items that are ready to use or preparing the items for use in-house.
Decontamination
Decontamination practices for aseptic processing must be documented in SOPs. This could include chemically sanitizing equipment to take into the cleanrooms, wiping items down with disinfectants, or using decontamination devices such as vaporized hydrogen peroxide (VHP) generators or autoclaves.
Facility Design
The design of the facility should be documented on maps and flow diagrams to help personnel in their daily tasks. The facility must be constructed to prevent microbial contamination. This could include items like differential pressure cascades, the use of classified areas, and pass throughs to name a few items.
There must be separate areas available for the storage and quarantine of materials. The warehouse must be neat, clean, and orderly, with temperature/humidity controls where appropriate. Cardboard or other items containing cellulose fibers should not be allowed in clean areas as they could be a source of mold contamination.
Laboratory practices must also be implemented to prevent microbial contamination from outside of cleanrooms. This could include changing uniforms and shoes and using proper aseptic gowning practices.
Automated or separative manufacturing designs such as isolators or restricted barrier access systems (RABS) can be utilized in the manufacturing areas to prevent cross contamination.
High efficiency particulate air (HEPA) and heating, ventilation, and air conditioning (HVAC) systems should be used with differential pressure cascades, temperature controls, and humidity controls to prevent microbial contamination. If the temperature is too hot or humid, people could sweat, compromising their cleanroom gowning. In addition, when pressure cascades are not controlled properly, microbes could enter the cleanrooms. Excessively humid environments can increase the potential for fungal contamination.
Effects from Adjacent Areas
The effects from adjacent areas should also be considered. If an adjoining room has microbial contamination, that contamination could migrate into the inner core of the cleanrooms. Transition areas should be monitored and controlled.
Supplier Qualifications
Qualifying suppliers is an important approach to control items that are purchased sterile and ready to use. It is important that vendors are trusted to provide quality supplies to maintain sterility assurance of the final product that is being manufactured. Supplier qualifications must be governed by SOPs.
Validation and Qualifications
Clean areas must be validated and maintained. This should include environmental monitoring qualification programs (EMPQ) and cleanroom qualifications. Equipment should also be qualified for use in the cleanrooms. There should be cleaning validations that include clean and dirty hold times of equipment.
Also, in the realm of validations and qualifications is the topic of process validations (i.e., media fills). Media fills help to demonstrate that the manufacturing process can produce a sterile final product. The manufacturing process should include interventions and aseptic connections. “An appropriate microbiological media will be treated as if it were product, and it will be put through an entire manufacturing process simulation. This study will show contamination control effectiveness throughout the manufacturing process.”4
Decontamination, Cleaning, and Disinfection Programs
Decontamination, cleaning, and disinfection programs must be established. The programs must be governed by SOPs and should describe what gets cleaned, how the cleaning is performed, how often the cleaning is performed, what cleaning agents are utilized, and the validation of the cleaning, decontamination, or disinfection process. Room cleanings should include items like the walls, floors, ceilings, and equipment. Again, there should be established clean and dirty hold times for equipment and the cleanrooms.
When utilizing disinfectants, consider items like disinfectant efficacy date, wet contact times, and the method of application of the disinfectants. Cleaning, disinfection, and/or decontamination concepts should be considered for both product contact and non-product contact surfaces.
Product and Materials
Products and materials must also be controlled to prevent contamination and increase sterility assurance. Raw materials, components, active pharmaceutical ingredients, container closures, and product contact surfaces should all be monitored and controlled. “Sterility must be assured for cleaning solutions, tools and equipment, raw materials, container closures, and any other materials that will be introduced into the area.”4
Manufacturing Practices
As previously mentioned with media fills, the manufacturing process must be monitored and controlled. This includes interventions and aseptic connections. Proper aseptic technique and behaviors must be utilized to prevent cross contamination of product during manufacturing.
Laboratory Testing
Laboratory testing to consider when thinking of sterility assurance programs include items like sterility testing, endotoxin testing, bioburden testing, raw material testing, in-process testing, and container closure integrity testing. “A sterile product is to undergo analysis for microbial endotoxins and sterility testing to assure the absence of contamination.”4 This is not an all-inclusive list of laboratory tests, but these quality control assays help boost the confidence in sterile products by providing a secondary layer to sterility assurance practices.
Environmental Monitoring Program
Environmental monitoring assesses the microbial contamination level in the cleanrooms and adjacent areas. This data may highlight areas that need extra cleaning, monitoring, and/or maintenance. Air and surfaces are routinely monitored within the cleanrooms to make sure the environment continuously meets specifications.4
Utility and Water Systems
Monitoring of utility systems includes water systems, compressed air, compressed nitrogen, compressed oxygen, or similar systems. Depending on the system, testing could include bioburden, total organic carbon (TOC), conductivity, or non-viable particulate sampling. When thinking of utilities, it is important to design facilities so that there are no drains or sinks in the cleanrooms as these can be a source of microbial contamination.
Storage Conditions
When considering storage conditions, it is important to think of warehouse cleanliness, order, and quarantine areas. Temperature and humidity should be monitored and controlled when required. Conditions should be maintained to ensure the sterility of the final product. In addition, container closure integrity should be established to ensure the product remains sterile in its packaging.
Conclusion
The components described above of a sterility assurance program for an aseptic manufactured product do not comprise an all-inclusive list. Rather, this list demonstrates the breadth of the items that could impact the sterility of a product or device that is purported to be sterile. When designing a sterility assurance program, consider all the items that could impact sterility and develop procedures and methods that will increase the assurance of the sterility of the product or device and minimize risks to consumers.
References
- United States Pharmacopeia (USP) <1211> Sterility Assurance. USP 43-NF38.
- AMSCO International (1995) A Guide to Sterility Assurance: Practical Concepts for Your Institution. AMSCO Sterility Assurance Products. Accessed on 30Sept2021 at https://research.unl.edu/events/docs/AGUIDETOSTERILITYASSURANCE.pdf
- FDA (2004) Guidance for Industry: Sterile Drug Products Produced by Aseptic Processing — Current Good Manufacturing Practice. Accessed on 30Sept2021 at https://www.fda.gov/media/71026/download
- Skowronek, Kristina (2019) Safe Drugs, Happy Consumers: Sterility Assurance Programs. American Association of Homeopathic Pharmacists. Accessed on 30Sept2021 at Safe Drugs, Happy Consumers: Sterility Assurance Programs - The American Association of Homeopathic Pharmacists (theaahp.org)
About The Author:
 Crystal M. Booth, M.M., is a senior technical director at PSC Biotech, with more than 20 years of experience in pharmaceutical microbiology, environmental monitoring, and quality assurance. She obtained her master’s degree in microbiology from North Carolina State University. Booth is an award-winning technical writer and author of Method Development and Validation for the Pharmaceutical Microbiologist. During her career working as a consultant and for CDMOs, she has worked in microbiology, consulting, quality assurance, R&D, and quality control laboratories. She has developed and validated numerous microbial methods and has worked with many different product types.
Crystal M. Booth, M.M., is a senior technical director at PSC Biotech, with more than 20 years of experience in pharmaceutical microbiology, environmental monitoring, and quality assurance. She obtained her master’s degree in microbiology from North Carolina State University. Booth is an award-winning technical writer and author of Method Development and Validation for the Pharmaceutical Microbiologist. During her career working as a consultant and for CDMOs, she has worked in microbiology, consulting, quality assurance, R&D, and quality control laboratories. She has developed and validated numerous microbial methods and has worked with many different product types.