
Strategies For Minimizing The Impact Of Bioburden And Sterility Testing On Gene Therapy Batch Yield
By BioPhorum

It is widely recognized that gene therapy manufacturing processes result in low yields, particularly in early product development stages. Gene therapy products, however, are subject to the same regulatory release criteria and expectations as processes used for biologics/pharma products that produce significantly higher yields. Often, if gene therapy manufacturers were to adhere to current release requirements, the outcome would be little, if any, remaining product for the clinic or the patient.
This article outlines strategies for reducing the volumes required for bioburden and sterility testing and, therefore, conserving product for patients while remaining compliant and delivering assay and process information on the microbiological status of gene therapy products.
Sample Sizes
Despite the potential of advanced therapy medicinal products (ATMPs) to vastly improve the lives of patients, unique and often inefficient (low yield) biological manufacturing processes present challenges to sponsors, e.g., compendial in-process and quality control testing required to ensure the safety, identity, strength, purity, and quality of each lot manufactured. Common bioanalytical quality control testing can be achieved using small sample sizes (<100 μL to 500 μL), whereas traditional microbiological testing uses many milliliters of sample to achieve reliable results.
Unlike traditional small molecule and biologics products, the typical bulk drug substance (BDS) and corresponding finished drug product (DP) lot sizes for ATMPs are commonly <1,000 mL and very often <100 mL. Considering the traditional compendial in-process control and lot release testing sample volume requirements, and the requirement to retain samples, the entirety of a lot produced could be consumed if strictly following the relevant compendia.
Alternative sampling and testing approaches are required to demonstrate the microbial safety profile of gene therapy products while working within the confines of a significantly reduced amount of final product compared to traditional pharmaceutical and biopharmaceutical products. However, due to their novel nature, little guidance exists to inform investigational new drug application sponsors on the potentially acceptable pathways for reduced sampling and testing.
Survey Results
We conducted two surveys on the approaches used by companies and the feasibilities/risks of alternative approaches to bioburden and sterility testing, focusing on gene therapy aspects only.
The headline results include:
- ~78% of respondents reported that the total BDS produced was ≤1,000 mL,
- the total lot size and fill volume of the formulated and filled DP corresponded well with the responses for BDS, with ~75% of respondents filling <500 vials per lot and ~93% filling <10 mL per vial, and
- ~33% of respondents were filling very low volumes (<1 mL) per vial, which has a significant impact on lot yield when coupled with a small lot size.
Bioburden
Current Strategies And Guidance
Bioburden testing methodology requirements stem from several harmonized compendial guidance documents. These include USP <61> Microbiological examination of non-sterile products: Microbial enumeration tests and Ph. Eur. 2.6.12 Microbiological examination of non-sterile products: Microbial enumeration tests.
While harmonizing these compendial texts in 2009 (ICH Q4B Annex 4A(R1)) was a major step in bringing together the routine microbial compendial release tests for traditional large-volume non-sterile DPs, the documents did not consider emerging ATMPs. With the recent acceleration of gene therapy research, development, and commercialization activities, many sponsors struggle to define a bioburden testing approach for these products within the confines of the published guidance.
The guidance documents remain silent on the bioburden testing strategy for a gene therapy manufacturing process and provide limited guidance on the appropriate limits and sampling volumes for both in-process and BDS testing.
Unique Gene Therapy Considerations, Challenges, And Recommendations
When designing a microbial monitoring plan for a product-specific or a platform manufacturing process, using a comprehensive contamination control strategy is essential in identifying risks and the current controls that may be implemented. The contamination control strategy will be a valuable tool and may help to justify when nonstandard sampling and testing approaches are taken.
While most harmonized compendial guidance does not apply to gene therapy products, the statements on testing active pharmaceutical substances can be applied to design a compliant BDS testing strategy using 1% of the total batch. Less than 1% of the final batch may also be tested but would require the sponsor to prepare a risk-based justification and seek approval from the relevant health authority.
Upstream Sampling Plan
Care should be taken to evaluate the contamination risk to the process and the potential impact on yield. The latter is generally negligible as a result of any upstream microbial sampling as the target vector is not removed from the process during sampling (only the cell culture used to produce the vector).
The upstream process relies on a near-sterile environment for successful cell culture expansion. Biological safety cabinets or isolators are used for small-scale cell culture manipulations/passaging, and larger-volume, single-use closed-system equipment is used for the later stages of the expansion/production process. Low levels of microbial contamination introduced during these stages of the production process can result in batch termination. Microbial sampling from culture intended to be forward processed is generally not recommended, except for the unprocessed bulk-harvest sample.
Evaluating any potential sampling plans should be commensurate with the risk to the process posed by the manufacturing process. Often, adherent cell culture production processes involve a larger number of manual manipulations compared to suspension culture processes. Consequently, microbial monitoring may be performed on high-risk operations, but the recommended approach is to sample from process wastes or residual cell culture volumes that remain after the operation.
Downstream Sampling Plan
Due to the open-system/partial open-system nature of downstream processing, a robust microbial monitoring plan is required to ensure bioburden control is maintained through these non-sterile operations. The downstream in-process and BDS sampling plan presents a potentially large impact on batch yield as the target vector being produced is removed from the process with each sample taken and the large reduction in batch size as the vector is concentrated and purified. A 500 L cell culture suspension at bulk harvest can yield as little as 100 mL to500 mL of BDS, which is ≤0.1% of the total starting volume.
Routine test samples plus backup/retain samples can quickly deplete available volumes and present a challenge to batch yield. Sampling volumes and volumes for associated backup/retain samples should be evaluated based on process and laboratory performance history. A minimum of double the required amount for testing is recommended for sampling, divided into one sample for the performance of the routine test and one backup sample. Using retain/backup samples should be evaluated carefully, particularly regarding sample hold times and storage conditions before testing. Without this data, in-process backup/retain samples will be of limited value, as the validity of the retest data cannot be confirmed if testing is performed more than 24 hours after the initial testing.
Bioburden Testing Of BDS And Final Formulated DP
By following the industry-standard bioburden sampling and testing volumes/limits presented by non-invasive micro-test technology (10 CFU/100 mL), 10%-100% of a batch may need to be tested for products that yield ≤1,000 mL. This provides potential justification for using a lower sample volume, primarily the use of one that is approximately 1% of the total batch volume.
Testing Methodology
The harmonized compendial guidance provides testing methodology advice for three options: membrane filtration, plate-count methods, and most-probable number method. Due to the aqueous nature of the samples collected from a typical cell culture biomanufacturing process, membrane filtration provides the simplest workflow for the quality control analyst and reduces sampling volumes by only requiring a single replicate for testing.
Method Suitability
Some strategies can be used to generate method suitability data while reducing the impact from sampling, including:
- using toxicology/product development/engineering run material that is not intended to be dosed to patients is a possible avenue in early development,
- use a “family approach” or worst-case scenarios from the process to group similar samples under one suitability test, and
- using placebo or buffer samples in place of the actual product.
Sterility
Current Strategies And Guidance
Sterility testing is performed on DP vials per compendia requirements USP <71>, Sterility Test and EP 2.6.1 Sterility. Testing requirements are based on two factors.
The Volume Of The Filled Vial
Table 1 shows the requirements for the number of vials to be tested based on filled vial volume.
Table 1: Sterility testing requirements based on filled vial volume
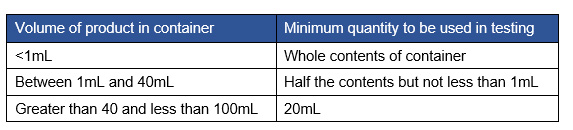
Of the survey respondents, ~30% indicated that fill volume is <1 mL, which would require additional vials to meet the 1 mL requirement for each media type. Note that any fill <2 mL would require additional vials to be pulled to meet this 1 mL volume requirement. The remainder of the sponsors would fall in the range of >2 mL and would meet the requirement for the 1 mL minimum for each media type.
The Number Of Vials Filled In A Respective Batch
Table 2 shows the requirement in the sterility chapter for the prescribed number of vials to use for the sterility test.
Table 2: Vial number requirement for sterility test
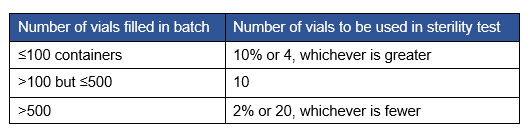
Per the compendia, if the contents of one container are enough to inoculate the two media (TSB, FTM), the number of vials column in Table 2 provides the number of containers needed for both media. Note that for volume sizes between 1 mL and 40 mL, the requirement is half but not less than 1 mL. This may require more vials for vial fill sizes that would not meet the 1 mL requirement for each media type. Sixty percent of respondents indicated that the number of vials required would fall in the >100 but ≤500 category, with a requirement for 10 vials.
Additional Gene Therapy Considerations
Suitability Testing
An additional requirement of sterility testing is to ensure that the method is suitable for use with the product. Here, the product is challenged with organisms to ensure that their growth can occur in the presence of the product. More volume of product would be required to perform this testing.
Some companies use post-incubation suitability testing. Direct inoculation testing for sterility may be performed as an option for small-volume testing. The recommendation would be for products under 10 mL.
When direct inoculation is performed, suitability testing could occur post-incubation of the routine testing, which would conserve material. This approach should be taken when there is a reasonable assurance that there would be no interference based on prior suitability testing or knowledge of possible interference of the matrix. If interference was shown after the testing was completed, the initial test would be invalid, which is a risk of this approach.
Variable Fill Volumes
One reported approach to reducing required volumes for testing is to have a “variable fill volume”. This would be accomplished by starting a product fill with a lower volume for a defined number of vials that would be used for product testing, and then continuing the run with the defined clinical fill volume. This would be inappropriate in cases where stratified sampling is required (typical with sterility) or where the fill volume is a consideration for testing. Based on the surveys, 40% of respondents reported using some type of modified fill volume. Approaches included using research vials at a lower volume than GMP and using lower fill volumes for analytical samples.
Conclusion
Significant reductions in the volume required for testing may be achieved compared to typical testing volume requirements in both bioburden and sterility testing for in-process, drug substance, and DP testing.
Approaches for organizations to consider include:
- leveraging existing verbiage in compendia for small-volume products,
- taking platform and phase-appropriate approaches,
- taking matrix-based approaches for suitability testing requirements,
- using placebo or buffer samples in place of the actual product, and
- using a reduced sampling plan.
This article summarizes a recent BioPhorum publication on the topic. To read more, check out the full paper, Minimizing the impact of bioburden and sterility testing on gene therapy batch yield.