
Risk-Based Strategies For Revalidating Bio/Pharma Equipment & Systems
By BioPhorum

Validation of equipment and systems is a regulatory requirement in the pharmaceutical industry, which must demonstrate that medicines are safe, effective, and of suitable quality. Ongoing conformance to operating parameters, alarm functions, cleanability, etc. (including when changes are made) is called “maintaining the validated state” and is a requirement for revalidation or a continuous monitoring control strategy.
To make a pharmaceutical product, numerous pieces of equipment and systems are required, including those that are direct product contact (e.g., fermenters) and those that are indirect (e.g., autoclaves). All equipment and systems are validated when first introduced to the facility and process. They have multiple operating parameters that are important to the manufacture, storage, handling, and testing of a pharmaceutical product.
However, it would take thousands of annual/biannual tests if all equipment and systems had to be retested and it is impossible to retest all those parameters initially tested during validation. But which should be tested to ensure the ongoing validated state? One area of focus should be those that can be defined as critical and high risk if they fail. Another is how frequently those critical parameters should be checked.
Parameters and process controls that are critical to the safety, quality, and efficacy of the products must be properly controlled every time the equipment or system is used. These may include:
- controls around critical process parameters (CPPs), e.g., concentration
- stability of the product during the process and storage
- impact of changes to the process, equipment, and systems.
Collaboration between those who understand the equipment and those who understand the process is essential. It is important to understand what elements are critical to the safety, efficacy, purity, and traceability of the product and separate these from the important parameters relative to how the equipment operates. This makes it possible to manage a compliant revalidation program or set up a continuous monitoring program that is manageable in this highly automated industry.
This article provides an overview of risk-based approaches to revalidation and assesses what is required to maintain the validated state of equipment and facilities. The approaches summarized below do not include all revalidation needs, but the same concepts can be extrapolated to other equipment, processes, cleaning, and systems.
New Product Introduction (NPI)
Some testing (or validation in commercial facilities) is required when new products are introduced. Multiple equipment and systems can be impacted by bringing in a single new product. However, science- and risk-based considerations should be used to define the testing required in these situations.
The strategy for any NPI can be defined in a validation plan, by change control, by protocol, or by procedure. The primary purpose of the evaluation is to describe the impact that the new process has on existing equipment and systems. It should also ensure that existing equipment and systems are suitable for each existing/new process and be able to defend that the existing/new product will be safe, effective, pure, and have a traceable history.
An evaluation would address questions such as what “elements” of the new product/process are CPPs or critical quality attributes and what is the impact of those on existing equipment and systems? And what potential impact might the changes have on current products/processes and what “evidence” will be required to defend that they are not impacted?
Performing a risk assessment is the most effective way to get from the NPI to the final determination that the equipment and systems are continuing to operate in a validated state. For example, the risk assessment might include these actions:
- Map out the changes required to the equipment, systems, and processes to introduce the new product.
- Define the potential impact these changes could have on the quality of both the proposed new product and existing products.
- Define the evidence that supports the current products and determine whether this evidence can also be applied to the new products.
Hold Times
These are used to define a maximum amount of time that processes/steps can be held, or must be completed within, to avoid negative impact on the microbial quality and/or chemical quality of the product. Once the maximum allowable time has been determined, it is established as a manufacturing control to maintain the validated state of the product/process. If established times are exceeded, the impact on quality and chemistry must be assessed.
When a manufacturing facility is at maximum capacity, finding time for at-scale testing for hold times is challenging. This often results in hold times driven by schedule limitations rather than quality limitations, which can significantly impact the flexibility and ongoing scheduling of manufacturing. In terms of risk, the purpose of assessing hold times is to define a time limit within which the product is not impacted.
Using a science- and risk-based approach, hold times can be applied to any process step. However, they are most commonly required for steps where the hold time is critical to the process (e.g., storage), where the hold time is critical to the microbial control of the product (e.g., pre-filtration media hold times), or where the hold time is critical to the chemical quality/stability of the product (e.g., viral inactivation at low pH in biologics).
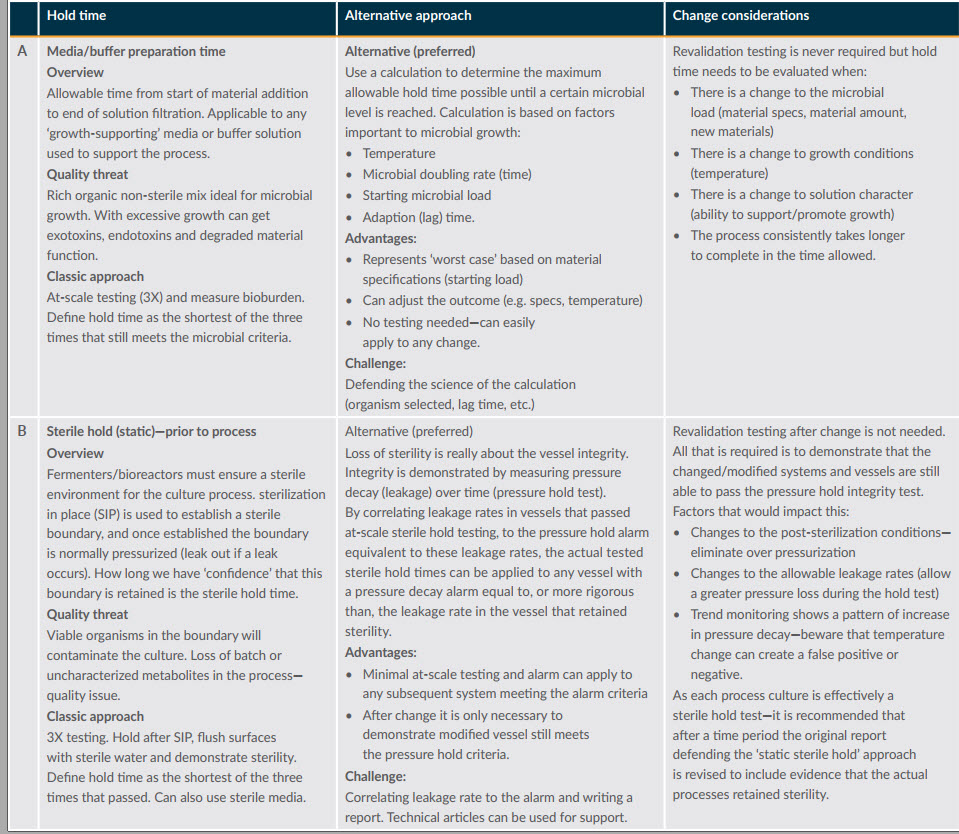
After defining the potential impacts, the next step is to think of alternative ways to gather evidence to defend the change. Figure 1 provides an overview of two simple processes for defending hold times.
Figure 1: Example hold time assessments
Controlled Temperature Chambers
Manufacturing facilities may have hundreds of controlled temperature environments. These may be single units or freezer farms and may include controlled warehouse environments and refrigerated and frozen cold-storage environments.
Storage outside the required conditions can directly impact quality. Chambers are typically validated to demonstrate that the environments used for the storage of materials and/or product can be controlled and maintained within the required ranges for the materials and products to be stored and to provide evidence that the monitoring probe(s) used give a reliable measure of this environment.
The risk assessment needs to focus on issues such as how critical the materials/products stored in the chamber are and how sensitive they are to temperature changes. These risks should be factored in when assessing the impact of changes and the risk to product quality and in defining testing and frequencies for maintaining the validated state of equipment. Remapping is still the major revalidation activity performed to confirm the validated state of the equipment.
This is where data can help support the justification. If there is data to support that a freezer maintains a controlled environment for months and years, this can be used to support defining the frequency of remapping. Revalidation data can also be used to determine if freezers have drifted after a year, and that data can be used to define a new frequency.
Depending on the continuous monitoring system/monitoring programs/plans, an automated system can be used with validation mapping inputs to identify temperature shifts at the point of use. The system could send automatic notifications for immediate evaluation and serve as a real-time monitoring system for critical temperature-controlled units.
Sterilization Processes And Equipment
Most companies must define strategies for validation and revalidation of equipment sterilization.
Microbial inactivation using moist heat, gases, chemicals, dry heat, or radiation fundamentally follows first-order reaction kinetics. The logarithmic rate that organisms are inactivated during the cycle is constant. While conditions are constant, the number of viable organisms remaining is based on the lethality of the cycle conditions (temperature and time).
Factors relevant to sterilization cycles include starting microbial load and resistance of the organism or spores to the proposed cycle. Factors that may reduce cycle effectiveness include dryness requirement and post-cycle controls and the integrity of the sterilized system/container after the cycle.
For an equipment item to be sterile, clear requirements must be met, as a failure of product sterility is a critical risk to patients. However, sterilization in place (SIP) is often used loosely and most processes are just “microbial control steps”. A risk-based approach is important and can save a significant amount of effort in the initial validation and the post-validation maintenance of changes.
If a cycle is defined as SIP-sterile, it requires strict testing and revalidation requirements. Time spent evaluating and defining the real requirements for each specific part and where it is used in the process will simplify the overall process for the cycle. A SIP-bioburden reduction would have to be validated the first time it is used but may not need subsequent revalidation or could be revalidated every five years. By defining specific SIP-sterile loads, confirming that cycles are equivalent, and using overkill cycles, revalidation of these loads (one per year) could be rotated on an annual basis.
Periodic Review
This is another type of risk review and is an element in the overall strategy for ensuring and defending the quality of products given to patients. The purpose is to ensure that systems, processes, and facility/equipment remain in a validated state of control and are suitable for use over time. Many factors are impacted by time/use, including wear/damage to mechanical parts and changes in function and performance of elastomers and seals.
The impact of the quality system processes over the period also needs to be assessed, including, for example:
- post-validation changes that have been made to the systems, equipment, and processes
- corrective maintenance work
- corrective actions from quality events, etc.
- number of exceptions and performance issues associated with the system.
A risk-based approach should be used to define the overall quality strategy for periodic review of all GxP systems and processes at a site, based on the threat level to patients. This strategy will assess the criticality of the system to product quality and the ability to detect performance degradation (e.g., continuous monitoring of water systems) and regulatory requirements (that cannot be “risked away”). The risk-based approach can also define whether the review can be of real-time information and data (e.g., automated systems) or process information based on the release data of batches made over a period. A risk assessment can also define what information is not required, such as not looking at change controls if your program ensures that, after a change, the equipment is placed back in the validated state.
Conclusion
Organizations understand their facilities, equipment, processes, and systems from a holistic perspective and can determine what works best for revalidation. The revalidation best practices summarized here are a collection of concepts and processes that have been used successfully across the industry. There may not be a one-size-fits-all approach to revalidation, but many of these practices can streamline and focus revalidation requirements on those that offer the most value and are not just testing and documentation for no positive gain. By defining the controls up front for equipment, relying on data, and using scientific understanding, an organization can set itself up with a simple, compliant, and valuable revalidation strategy.
This article is a summary of a recent BioPhorum publication on the topic. To read more, check out the full report in Risk-based strategies to support revalidation and the assessment of requirements to maintain the validated state of equipment, process and facilities used for commercial and clinical manufacturing