
Regulatory Concerns For Facilities Of The Future: Does Flexibility Come With A Cost?

By Eric Langer, president and managing partner, BioPlan Associates, Inc.

Trends in biomanufacturing follow a slow cycle, partly due to the regulated nature of the industry and the substantial costs and risks that introducing a change in the process can bring. But that ignores the benefits that changes in bioprocessing over the past 10 years have brought, from better expression systems, to new modular and flexible facilities, sensors, and control systems for downstream operations.
Single-Use Regulatory Concerns
One area that has brought significant change is disposable/single use bioprocessing devices. Regulatory concerns are increasing significantly as these plastic devices are brought into clinical, and even commercial applications. When we asked end-users in our 12th Annual Report and Survey of Biomanufacturing about critical reasons for not increasing the use of disposable technologies, two of the top four factors are regulatory-related. The second-most critical concern this year, “For regulatory reasons, we can’t change our current systems,” moved all the way up to #2, from the 10th spot last year, with a big jump from 2.2 percent to 15.2 percent of respondents. This is an indication that these novel bioprocessing devices are becoming fully mainstream, and the need to address previously theoretical regulatory factors is now more pressing.
As most end users determine that single-use systems can provide benefits over fixed stainless steel systems, the regulatory concerns will likely increase. The third most common issue was leachables and extractables concerns. These are also a regulatory issue, and are worrisome because of what could potentially be leached or extracted out of the plastic bags, fittings, and films during bioprocessing operations. Most single-use contact surfaces require evaluation for regulatory purposes to ensure they are safe for contact with the drug product.
Fig. 1: Top Reasons For Not Adopting Single-Use/Disposable Technologies
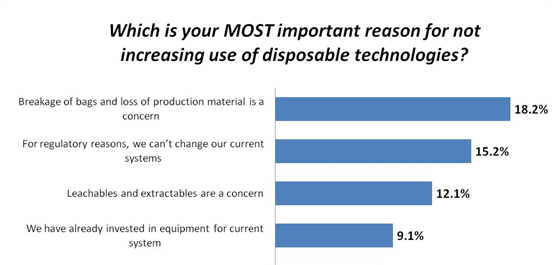
Data source: 12th Annual Report and Survey of Biopharmaceutical Manufacturing Capacity and Production, www.bioplanassociates.com/12th
Hiring Regulatory Staff
The concern over regulatory issues is a consistent problem. Over the next 12 months, according to our study, biotherapeutic developers and CMOs plan to bring onboard a variety of new staff. Of these, 34 percent are expected to be involved in “Production operations,” yet “Regulatory/QA/QC” department growth will remain steady at 14 percent.
Fig 11.1: New Hires In Biopharmaceutical Manufacturing (2015)
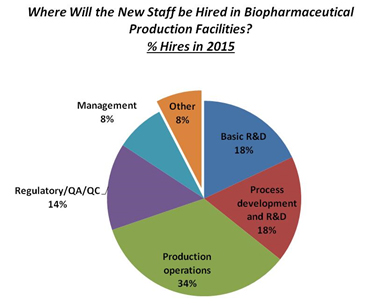
Data source: 12th Annual Report and Survey of Biopharmaceutical Manufacturing Capacity and Production, www.bioplanassociates.com/12th
Projections regarding where hiring will take place over the next five years show staffing will largely occur in the same areas as present; hiring in “Regulatory/QA/QC” departments over the next five years will remain virtually unchanged. Overall, projected ‘5-year’ hiring trends over the past seven years have remained relatively stable.
International Regulatory Factors Creating Capacity Constraints
Although regulatory issues remain critical in the adoption of new technologies, respondents to the study were also concerned about major factors expected to constrain their organization’s production capacity over the next five years. The most frequently indicated factor was “Facility constraints” (60.1 percent, up from 52.4 percent last year). But Inability to meet international regulatory requirements was noted by 11.5 percent of respondents as a factor constraining capacity. This international concern is contrasted with the lowest-reported factor at 5.4 percent, “Inability to meet domestic regulatory requirements in a timely manner”. Thus, while international regulatory problems may impact capacity, general domestic regulatory considerations remain limited.
On the other hand, when we split out two groups of end-users, biotherapeutic developers and the industry’s contract manufacturing organizations (CMOs), we find significantly different views on future capacity constraints. This year, CMOs again expressed higher levels of concerns for “Lack of regulatory staff” (21.1 percent vs. 9.3 percent for developers). Clearly, CMOs see their regulatory staff as a very substantial problem compared with their in-house counterparts.
Over the past few years, respondents have more frequently expressed a desire for standardized international regulatory processes and increased training and education. Although the need to “Streamline FDA regulatory process” decreased from 38.2 percent to 32.9 percent, this year, it remains a concern for nearly a third of the industry. This includes many companies still waiting for further guidance from the FDA concerning biosimilars approvals, although some advances have been made, with finalization of some biosimilars guidelines in early-mid 2015.
Continuous Bioprocessing And Regulatory Problems
Another area where regulatory concerns are inhibiting innovation appears to be in the adoption of continuous bioprocessing (CBP) technologies. The classic approach to bioprocessing, both upstream and downstream, remains batch processing, which remains dominant, particularly at larger and commercial scales. But a number of technological advances are making continuous bioprocessing attractive. Continuous bioprocessing strategies are being adopted or considered for many new drug bioprocesses, and some established bioprocessing systems are being retrofitted and upgraded for more continuous operations.
There are many benefits to operating bioprocesses continuously rather than in batch mode, including:
- Reduced costs
- Increased productivity
- Improved quality
- Increased flexibility
However, current manufacturers and vendors are inhibited by a number of factors, including problems in changing existing production lines once introduced, the perceived complexity and cost of implementing a CBP technology, and the lack of trained operators. Further, regulatory expertise is lacking in this area, both at the end-user level and with the regulators themselves. In a separate study, GMP issues figured very prominently as a concern in the adoption of CBP.
Other Regulatory Hurdles Impacting Innovation
Membrane chromatography in commercial manufacturing offers advantages when used to produce clinical material. There are some additional advantages that can be realized in late-phase and commercial production. Validation can be simplified and costs can be reduced through the elimination of several costly and extensive studies normally associated with diffusive chromatography beds. But regulatory factors come into play.
Single-use bioprocessing equipment, generally composed of plastics, now thoroughly (≥85 percent) dominates the smaller pre-commercial bioprocessing market. The market for single-use equipment will grow rapidly as regulatory issues around testing and documentation, i.e., polymers/plastics and fabricated equipment source materials and processing, and materials testing procedures are adopted. Tests and documentation appropriate for bioprocessing and regulatory submissions are being developed, including the polymer/plastics manufacturer, providing more basic extractables testing data, and related assessments of potential toxicities and contamination of bioprocessing and its end-products, including patient-relevant safety assessments. Although few polymer/plastics primary manufacturers provide exhaustive product toxicology studies and safety assessments, this is changing. The need to satisfy FDA and other regulators, full characterization, understanding, and, particularly, supply chain consistency is required with the materials and equipment used for bioprocessing. Success in this market also requires greater transparency, with polymer/plastics manufacturers (and equipment manufacturers) providing basic manufacturing, testing, and supply chain data as they work with bioprocessing suppliers and end-users to provide a steady, fully reliable, documented supply chain.
References:
- 12th Annual Report and Survey of Biopharmaceutical Manufacturing Capacity and Production, April 2015, Rockville, MD www.bioplanassociates.com/12th
About the Author:
Eric S. Langer is president and managing partner at BioPlan Associates, Inc., a biotechnology and life sciences marketing research and publishing firm established in Rockville, MD in 1989. He is editor of numerous studies, including “Biopharmaceutical Technology in China,” “Advances in Large-scale Biopharmaceutical Manufacturing”, and many other industry reports. elanger@bioplanassociates.com 301-921-5979. www.bioplanassociates.com
Survey Methodology: The 2015 Twelfth Annual Report and Survey of Biopharmaceutical Manufacturing Capacity and Production yields a composite view and trend analysis from 237 responsible individuals at biopharmaceutical manufacturers and contract manufacturing organizations (CMOs) in 28 countries. The methodology also included 164 direct suppliers of materials, services, and equipment to this industry. This year's study covers such issues as: new product needs, facility budget changes, current capacity, future capacity constraints, expansions, use of disposables, trends and budgets in disposables, trends in downstream purification, quality management and control, hiring issues, and employment. The quantitative trend analysis provides details and comparisons of production by biotherapeutic developers and CMOs. It also evaluates trends over time, and assesses differences in the world's major markets in the U.S. and Europe.