
Proposed Best Practices For Cell & Gene Therapy Aseptic Process Simulation
By BioPhorum

Aseptic process simulation (APS) plays a critical role in demonstrating that the processes, equipment, and materials involved in sterile manufacturing (e.g., facility/air-handling units, gowning procedures, and personnel) work in synergy to maintain sterility.
The regulatory and good manufacturing practice (GMP) requirements for APS of aseptically filled conventional drug products (i.e., traditional biologics such as monoclonal antibodies (mAbs) and enzyme replacement therapies) are well established through guidelines and historical experiences of aseptic manufacture, FDA warning letters, and 483 observations. It is well established that before the use of a new (or significantly changed) aseptic process in GMP manufacturing for drug product for human use, typically a minimum of three consecutive and successful APS runs are carried out to demonstrate process robustness and reproducibility.
APS runs, while representing a production process, are intended to challenge aseptic portions of the process activities under worst-case conditions. Traditionally, operators’ participation in APS execution is required as part of their initial qualification. After this, operators must participate in an APS execution a minimum of once per year thereafter to maintain their qualified status. For manual operations (e.g., aseptic compounding or filling), APS should be re-executed approximately every six months for each operator.
Applying existing APS guidelines to advanced therapy medicinal products (ATMPs) can be challenging since ATMP processes often differ significantly from those for traditional biologics.
This article explores the regulatory landscape and reviews and identifies challenges in implementing APS for ATMP processes. It proposes recommendations to guide industry in establishing viable and comprehensive APS programs with patient safety and regulatory compliance of paramount importance.
Existing Regulatory Guidance Landscape
In addition to regulatory and industry guidance, which primarily focuses on APS for conventional drug product filling, there are some specific information sources on approaches to APS for ATMPs. Table 1 provides a summary (extract only) of each regulation or guidance and highlights how it either supports the APS approach for ATMPs space or provides challenges.
Table 1: Regulatory landscape review (extract only). Click on image to enlarge.

APS Challenges In ATMP Processes
It is important to reemphasize that existing global regulatory and industry guidance for APS is mainly based on conventional drug products, with a focus on liquid and lyophilized fill and finish. Historical knowledge and experience with APS design for conventional drug products, although extensive, do not necessarily support the development of the strategic approach and design of programs for ATMPs, as production processes for ATMPs are intrinsically different from those of conventional drug products. In general, ATMP production processes can be categorized into three groups (see Table 2).
Table 2: ATMP production process category examples. Click on image to enlarge.
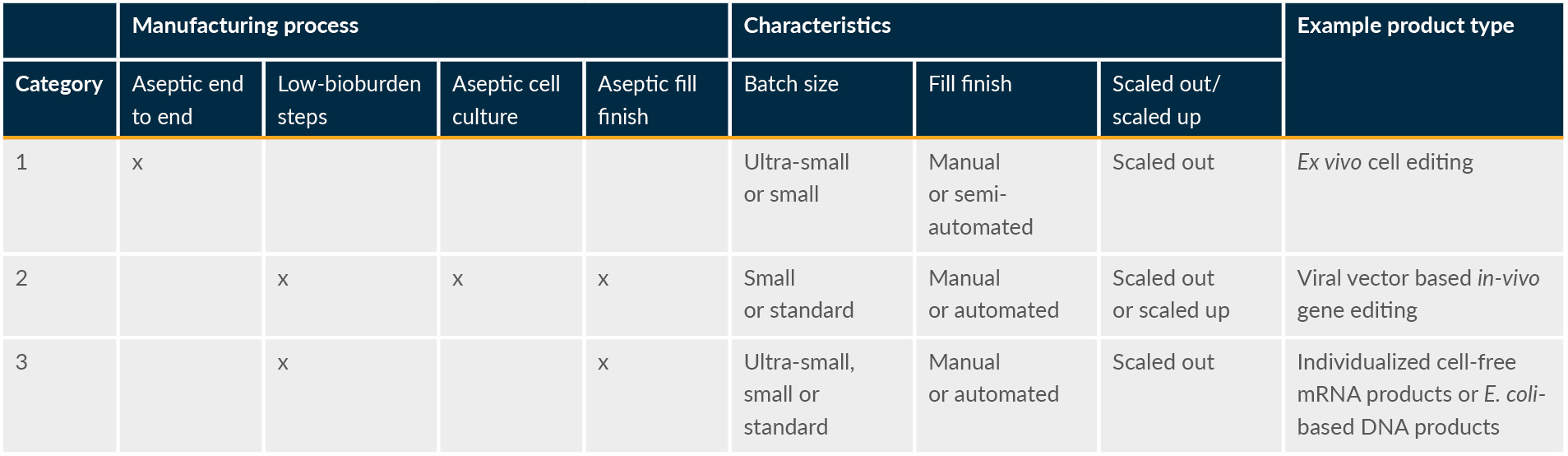
As shown in Table 2, ATMP manufacturing processes are diverse. However, there are shared key differences compared to manufacturing of conventional drugs, including:
- commonly, ultra-small batch sizes (can be as few as one or two units),
- highly variable, operator-dependent, and manual aseptic processes, and
- very lengthy processes (sometimes >30 days due to successive open cell and tissue processing steps) requiring asepsis to be maintained throughout with no opportunity to filter/sterilize the downstream product.
These differences result in different levels of complexity when applying current regulatory guidance on APS design to ATMPs.
What makes developing APS programs for ATMPs even more complicated is the limited acquired “know-how” due to the relatively short history of ATMP production with only a few licensed products. Moreover, guidelines on APS design for ATMPs are not well established. Therefore, there is a need for approaches to how APS requirements can be met practically in the ATMP sector while ensuring patient safety is not compromised.
Facility Readiness Before APS Work
The facility should be qualified before APS work by performing an environmental monitoring performance qualification (EMPQ). Here is an outline of requirements before starting the EMPQ:
- All major equipment, tables, and material storage are in designated locations.
- Air-handling units should be qualified according to grade classification.
- The maximum number of employees that will be required during production has been established, including manufacturing, quality assurance, and other staffing.
- A scheme for rotation of staff is established to ensure shift changes are adequately simulated.
- Routine room monitoring sample locations have been identified, justified, and established.
- All participants in the EMPQ have passed an aseptic gowning qualification; this ensures any failures are not due to improper gowning technique of the participants.
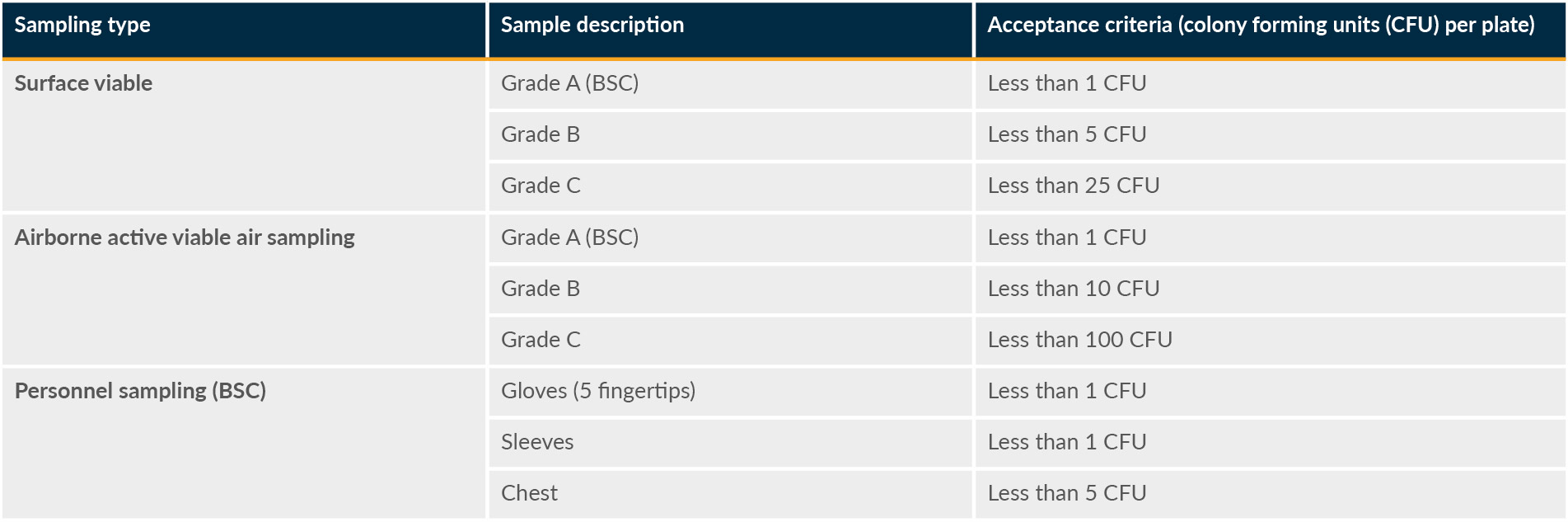
There are two qualification events for the facility: an at-rest EMPQ and an in-operation EMPQ. The at-rest EMPQ is performed while there are no employees in the cleanrooms for a specified time period. The in-process EMPQ is performed with the maximum number of employees necessary for the production in the cleanrooms for a specified time period. Table 3 summarizes the acceptance criteria for EMPQ sampling for static conditions.
Table 3: EMPQ acceptance criteria. BSC is “biological safety cabinet.” Click on image to enlarge.
Microbial Risk Assessment
A microbial risk assessment — which includes subject matter experts from quality assurance, quality control, manufacturing, facility, validation, process development, and other functions as required — may help determine potential contamination risks to the product from a number of sources. Where the risk assessment determines there to be no/low risk to the process, justification in the APS protocol may provide a rationale to adjust the APS design accordingly.
Considerations For Operator Qualification
All personnel authorized to enter the aseptic processing room during manufacturing will require qualification via participation in an APS representing the operator’s duties during routine production. However, given the high number of operators necessary in a scale-out approach and limited APS participation opportunities due to process family and equipment bracketing approaches, a formal plan for operator qualification could be considered. For open cell and gene therapy processes, there are three tiers of qualification: Grade A operators, Grade B operators, and ancillary staff. For closed processes, the aseptic risk is much lower and would not need to include operator qualification. Accordingly, the operator qualification (operator-specific APS) does not need to challenge closed interventions. Operators must still be fully trained in closed system interventions before qualification.
The operator-specific APS is an acceptable alternative to process-specific APS in terms of a positive qualification of each operator without the risk of jeopardizing the aseptic qualification of the process, equipment, and/or facility in case of a failure, as an operator-specific APS is typically executed offline without the use of patient material.
In the operator-specific APS, each technician must perform an initial aseptic qualification, which requires three successful manufacturing simulations, before becoming a qualified Grade A operator.
Once qualified, to maintain qualified status, the operator is required to complete an annual requalification. Requalification may require successful execution of the step that involves the most frequent and high-risk open manipulations.
Grade B operators must also be qualified in an APS based on their duties during routine production. The level of qualification for a Grade B operator should be based on the risk of that operator’s duties. Grade B operators must be qualified before participating as an operator on a GMP batch and annually requalified.
For functions that impact Grade A, the ancillary staff would typically have to participate in an APS before entering the room during GMP batch production. However, for staff who stay well segregated from the Grade A operations (e.g., observers, maintenance, engineers, quality control, quality assurance, and outside agencies), it is allowable to enter the room after completing gown qualification without participation in an APS.
Identifying Worst-Case Parameters (Bracketing)
APS should be performed for aseptically produced cell and gene therapy bulk drug substances, drug products, and any other type of products that are not terminally sterilized or filtered. Given that not every product combination can be assessed in a multiproduct facility, some assessment criteria are required to select the worst-case run parameters and will be summarized in a risk assessment document.
Table 4 highlights representative examples of worst-case operations and inherent interventions with processes divided into several unit operations. Worst-case unit operations were identified based on their sterility assurance risk. Worst-case operations would typically be open operations as these provide the greatest risk of introducing contaminants into the system due to some combination of manual intervention, duration, and exposure to the background environment.
Table 4: Example worst-case unit operations. Click on image to enlarge.

APS executions must include the maximum processing duration of open operations experienced during routine production and any high-risk aseptic manipulation that may be necessary during the manufacturing process. “Unplanned events” within aseptic practices, such as intentionally disrupting the laminar airflow in the Grade A environment, stopping the heating, ventilation and air conditioning system to the suite and biological safety cabinet, etc., should not be confused with worst-case conditions.
When identifying worst-case unit operations, the following general points should be considered:
- Testing should bracket the longest open process duration period.
- It is assumed that manufacturer flow kits and tubing welds are qualified by the vendor and therefore are not repeated during the APS. In-process tube welds are tested as part of the inherent interventions.
- Multiple/various types of containers are used.
- The maximum number of people required in the manufacturing area to perform the activity are present.
- If the same product is manufactured in various disconnected rooms that are not of a similar design and layout, three runs of APS will be executed for each room.
- If the same product is manufactured in various rooms that are connected (e.g., by a corridor of the same cleanroom classification) and alike in design and layout, a bracketing model covering this room may apply, i.e., if multiple rooms are equivalent in equipment and design, the initial APS can be performed (n=3) for one room and (n=1) for the subsequent room(s) if appropriately captured in a risk assessment.
Conclusion
For APS design for ATMP manufacturing, we propose applying existing APS guidelines and APS best practices based on risk. The proposed strategy is based on three key considerations:
- ATMP manufacturing processes are extremely diverse. While some of them are more like traditional manufacturing of parenteral drugs, others are significantly different.
- Existing APS health authority guidelines were developed for traditional manufacturing of parenteral drugs. Therefore, they do not necessarily reflect the specific needs of ATMP manufacturing.
- While requiring adopted approaches for the practical execution of APS in ATMP manufacturing, the intended purpose of APS studies, i.e., simulating and challenging associated risks, will be maintained as guiding principles.
The ATMP process categories outlined in Table 2 simplify mapping any given ATMP process against existing APS guidance to identify areas where process-specific solutions must be developed because existing guidance is challenging when applied to ATMPs. One key area is the development of operator-specific qualifications since many ATMP processes are still highly manual.
Developing APS protocols for ATMP manufacturing following quality risk management (QRM) principles is aligned with the recent update of EU Annex 1 Manufacture of Sterile Medicinal Products, which encourages manufacturers to apply QRM principles for aseptic processing and is consistent with other regulations globally. Given the diversity of ATMP manufacturing processes, it cannot and should not be expected that health authorities will publish targeted guidance soon. Still, they are expected to provide reasonable latitude to manufacturers by further emphasizing that manufacturers should develop their own QRM-based approach. Ongoing efforts in advancing and implementing automation, robotics, closed systems, and isolators in ATMP processes could help alleviate some of the challenges with APS design.
This article summarizes a recent BioPhorum publication on the topic. To read more, check out the full paper in Cell and gene therapy aseptic process simulation reflections.