
PIC/S Annex Update: What Is Your ATMP Control Strategy?

As of May 1, 2021 the Pharmaceutical Inspection Co-operation Scheme (PIC/S) has revised Annex 2 of its GMP guide addressing the manufacturing of advanced therapy medicinal products (ATMPs) to be sub-divided into two sections: 2A Manufacture of Advanced Therapy Medicinal Products, and 2B Manufacture of Biological Medicinal Substances and Products.1 While this guide is not an FDA or EMA guidance, it represents the consensus thoughts of a group that those regulators participate in. The guidance shares a similar flow and organization with the EU EudraLex Volume 4 Guidelines on Good Manufacturing Practice specific to Advanced Therapy Medicinal Products. Since the publishing of Volume 4 there has been an evolution of technology and deeper understanding by the regulators.
Scope Of Annex 2A
One of the major discussion points during the industry review of the draft Annex 2A guidance in 2019 was around what portions of the ATMP process fall under this guidance. Volume 4 describes a risk-based approach, and the FDA has issued guidance documents targeting clarification of what falls under which regulation. In an effort to write a consensus document for different nations, the delineation between medical practice and manufacturing was referred back to national law. Gene therapies’ targeted personalized application to an individual’s particular genome was another novel approach where heavy reliance on diagnostic testing (covered under other guidance) confounded the start of manufacturing. Ultimately, example flowcharts of processes were the most useful in describing and assessing the risk, similar to those used in traditional biotech. Clarification around plasmid vs. viral vector manufacturing and the addition of example processes in Figures 1 to 3 (see below) were changes made to the draft guidance to help address the scope.
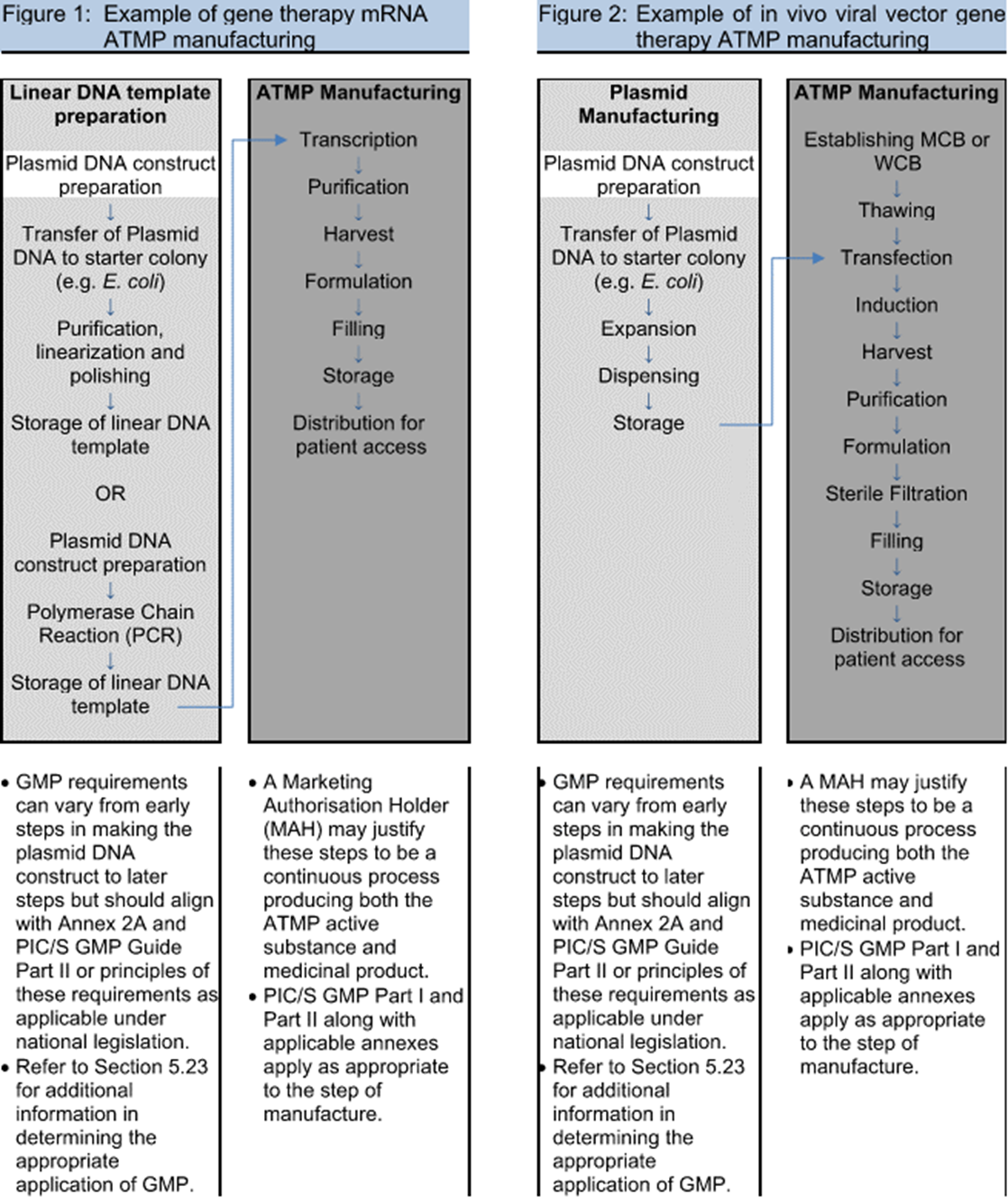
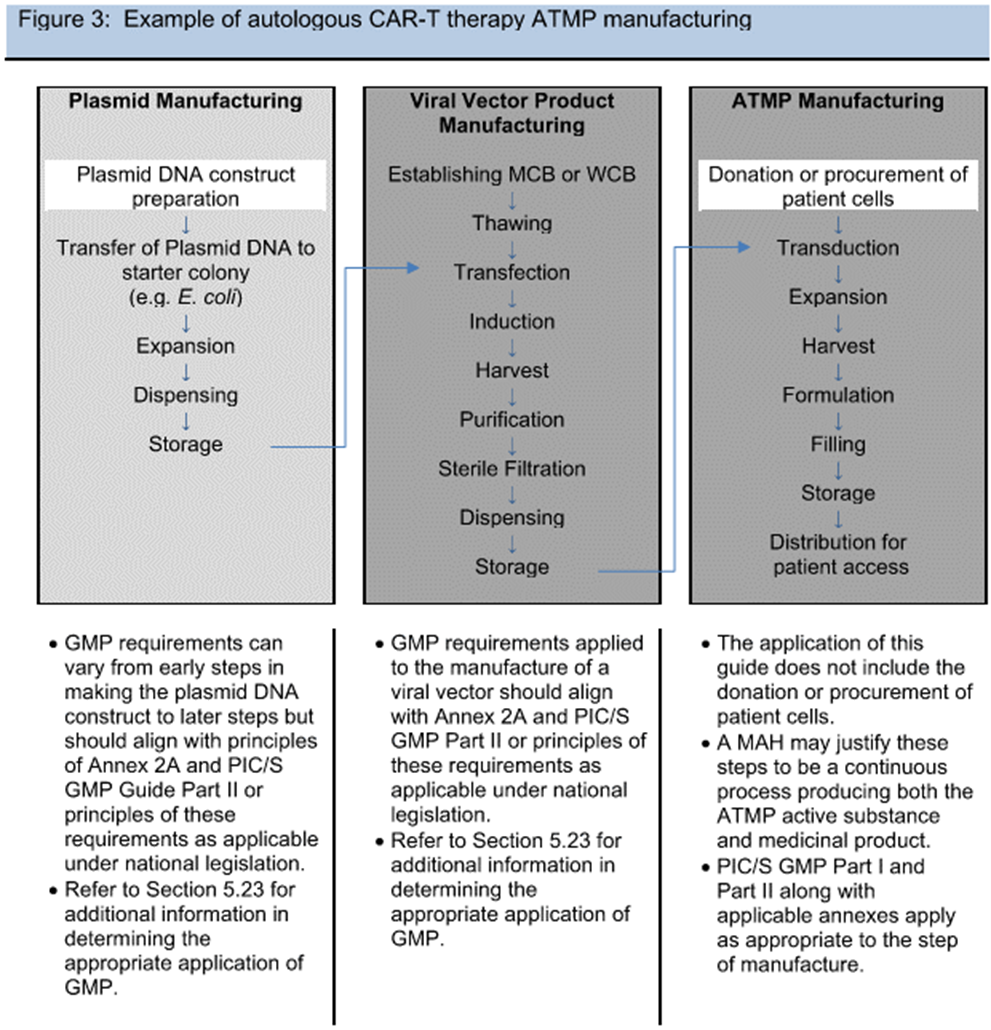
Figures 1-3 from PIC/S Annex 2A © PIC/S
As a result, viral vector manufacturing now clearly lies within the scope of GMP manufacture (dark gray background in figures), while plasmids are on a sliding scale of applicability to the GMPs (white to light gray background in figures), with construct preparation outside of GMP and increasing level of controls as the plasmid nears drug substance. Also, initial cell donation and isolation may fall under different regulatory guidance (varying by nation), but the handling practices and received material logistics need to be risk assessed. With all these variables and the unknowns of emerging novel therapies, process/product understanding and quality risk management are paramount.

Critical Aspects For Consideration Around Contamination Control
Contamination is a major focus of the Annex. How to develop a contamination control strategy (CCS) based on quality risk management (QRM) is not explicitly stated but points to consider are on nearly every page. ATMPs differ from traditional products on several major contamination control points: the potential lifesaving nature, the risk of migration of viral vectors from the intended step, patient to patient contamination, and the reduced ability to remove contaminants.
Due to the desire to maintain flexibility and limit resource expenditure, the industry continues to utilize biological safety cabinets (BSCs) and manual operations during manufacturing of clinical trial material. This differs from the regulatory desire to have a Grade A space with a Grade B background for aseptic manipulations; however, in Annex 2A an exception allows QRM-based justification for a less stringent environment for therapies treating life threatening conditions, subject to the regulatory authority’s approval. Some of the draft review debate highlighted the importance of research hospitals having the ability to manufacture trial material under good environmental control that aligned with medical practice but not the sterile products manufacturing requirements of Annex 1. The exceptional approval comes with a requirement for the manufacturer to improve the environment to GMP aseptic levels or to close the process as the technology is developed. Although we often hear the term “phase appropriate,” this may not be the correct approach. A serious risk assessment looking at the HVAC air classifications, manipulation processes with/without single-use systems, and the transfer systems must be made and contrasted with Annex 1, developing the risk to the patient.
Whether the clinical manufacturing facility is a research hospital or an isolated GMP suite at a biotech, the CCS is not just a “how to” on controlling today’s processes but also a path to mitigate risk as the therapy is scaled. Developing a transition plan to move from a manual process in BSCs to an automated/closed system over the course of manufacturing for clinical trials becomes critical and necessary to satisfy this expectation. This development strategy (timeline and comparability) needs to be discussed at the time of the initial application with the regulators.
In discussing production areas, the Annex highlights the risk of cross contamination of replication competent vectors. The FDA has shown diligence in identifying waste/raw material cross flows, both in facility design reviews and in recent enforcement action, for viral facilities. At clinical scale, spatial and temporal (time based) segregation might be accomplished; however, a commercial-scale facility will not be able to mitigate this contamination risk without physical segregation of flows. One notable exception to a CCS based on QRM is the defined expectation that for rooms with multiple isolators processing different vectors, the room needs 100% air exhaustion and unidirectional waste handling.
Viral risks extend beyond the vectors; a virally infected donor is explicitly stated as a risk to be considered. In clinical trials, it may be possible for participants to be screened and those with infections (e.g., HIV, hepatitis) to be excluded, but in commercial manufacturing, this needs to be addressed in a contamination control strategy, because the contaminated individual maybe the targeted patient, thus triggering the need to meet biosafety regulations for potential infectious material.
In section 3.15 there is a significant difference/specificity from normal biological drug substance guidance2 — the need to evaluate particulates due to the lack of filtration within many cell-based ATMPS. Due to the lack of process filtration, the impact of particulates from the single systems needs to be assessed and, if possible, a protocol developed to purge, sterilize, and prepare each single-use system (SUS) for use.
A validated system that enables the bidirectional tracking of cells/tissues from the point of donation, through manufacturing, to delivery of the finished product to the recipient is required per the Annex, including for clinical trial batches. Managing the chain of custody and identity is not a current regulatory requirement for non-cellular therapies. From cell donation through therapy delivery, the cells are to be labeled with all relevant information for traceability. This could be printed text for initial trials, but a 2D or 1D bar code, RFID, or equivalent mechanism will need to be implemented for any commercial production. A specific point of concern is with retrieval of cryopreserved material. Frost can obscure the label, and time out of the freezer needs to be minimized. Systems to track units to locations and non-visual identification (e.g., RFID) or robotically controlled material retrieval systems could mitigate this risk.
While similar to serialization in building a product history, there are unique challenges, particularly when working with multiple hospital systems and HIPPA personal health information. Approaches will vary between allogenic, donor matched allogenic, and autologous therapies. For autologous therapies, dynamic finite level manufacturing planning with the physicians on a patient-by-patient basis needs to be addressed to comply with the Annex. Tracking can be done manually, but this represents a risk that the FDA has pushed manufacturers to address with an electronic system before granting a BLA.
Validation & Quality
For Phase 1 and 2 trials, validation can be of a limited nature appropriate for the therapy based upon risk assessment. Regardless, aseptic simulations and validation of disinfection methods are expected per the Annex. Surrogate cells can be used for process validation of autologous or donor matched allogenic therapies with justification.
Written agreements are expected with each hospital supplying cells, as noted in sections 5.27 to 5.29 of the Annex. These sections set the expectation that the agreement will define the responsible parties and practices covering procurement of materials, handling of data, transport of materials, testing, and acceptance. The current CAR-T manufacturers have had to audit hospitals for compliance to quality standards as they would any other supplier. For clinical trials, selecting center(s) with experience in GMP manufacturing or administration of approved autologous therapies would be preferable.
The Annex states, “For many cell-based products, there is variability introduced through the starting materials that cannot be overcome by the manufacturing process or In-Process Controls (IPCs).”3 This variability is a major risk when setting specifications and manufacturing parameters. For example, Novartis produced a significant quantity of out of specification (OOS) material because the release specifications were set based on healthy donor variability not representative of the patient population.4
Due to the potential lifesaving nature of a product even if it fails to meet release criteria, effort should be made to make the product available to the treating physician, while outlining the potential risks. This is primarily applicable to autologous or donor-matched allogenic therapies where there may only be a limited window to treat the patient. The Annex indicates that procedures need to be in place to describe the steps for communicating the out of specification results, performing the risk assessment, and enabling case-by-case release to the treating physician.
Many of the cell-based ATMPs have expedited process turnarounds due to patient need or product stability. For those products, normal analytical tests (e.g., sterility, endotoxin) may not permit batch certification prior to administration. Alternative methods to obtain equivalent data are needed (i.e., rapid sterility test). This may lead to a two-stage batch certification. The initial certification releases the product for shipment and is based on in-process data, resolution of deviations, and rapid tests. The secondary certification is after the final test results have been completed. A procedure needs to be in place to address OOS results during the secondary review, including consultation with the treating physician.
These approaches will require reviewing batch records in short time periods or in real time. This will require new methods and technologies to manage, such as electronic batch records and manufacturing execution software, and their integration with laboratory information management systems (LIMS) and a deviation tracking system. These systems might need AI-based analysis to sort out deviations and identify critical ones for quick or online resolution and approval.
Conclusion
ATMPs bring new challenges that will require adaptation of good manufacturing practices. Annex 2A highlights unique aspects to consider around contamination control, traceability, and quality systems. With a few exceptions, the guidance is largely based on gaining an understanding of the process and controlling it through QRM.
References
- PIC/s, “Revision of PIC/S GMP Guide (PE 009-15),” https://picscheme.org/en/news.
- PIC/S, “Guide to Good Manufacturing Practice for Medicinal Products Part 1”, page 16
- Annex 2A page 24
- https://www.biopharmadive.com/news/in-car-t-manufacturing-a-hurdle-novartis-has-yet-to-clear/543624/
About The Authors:
 Herman Bozenhardt has 45 years of experience in pharmaceutical, biotechnology, and medical device manufacturing, engineering, and compliance. He is a recognized expert in the area of aseptic filling facilities and systems and has extensive experience in the manufacture of therapeutic biologicals and vaccines. He has a B.S. in chemical engineering and an M.S. in system engineering, both from the Polytechnic Institute of Brooklyn. He can be reached via email at hermanbozenhardt@gmail.com and on LinkedIn.
Herman Bozenhardt has 45 years of experience in pharmaceutical, biotechnology, and medical device manufacturing, engineering, and compliance. He is a recognized expert in the area of aseptic filling facilities and systems and has extensive experience in the manufacture of therapeutic biologicals and vaccines. He has a B.S. in chemical engineering and an M.S. in system engineering, both from the Polytechnic Institute of Brooklyn. He can be reached via email at hermanbozenhardt@gmail.com and on LinkedIn.
 Erich Bozenhardt, PE, is a lead process engineer for regenerative medicine operations. He has 15 years of experience in the biotechnology and aseptic processing business and has led several biological manufacturing projects, including cell therapies, mammalian cell culture, and novel delivery systems. He has a B.S. in chemical engineering and an MBA, both from the University of Delaware. He can be reached via email at erichbozenhardt@gmail.com and on LinkedIn.
Erich Bozenhardt, PE, is a lead process engineer for regenerative medicine operations. He has 15 years of experience in the biotechnology and aseptic processing business and has led several biological manufacturing projects, including cell therapies, mammalian cell culture, and novel delivery systems. He has a B.S. in chemical engineering and an MBA, both from the University of Delaware. He can be reached via email at erichbozenhardt@gmail.com and on LinkedIn.