
Pharma Quality Systems Need Revamping To Accommodate Digital Health Tech

As pharmaceutical companies develop and market more medical devices and combination products with digital health components, the FDA and industry experts are cautioning that the associated frequent software changes, software maintenance, and cybersecurity can carry significant regulatory and business risks.
In addition, bringing software into a pharma quality system has challenges similar to those that companies face when moving into the medical device arena, as the language, regulation, development timelines, and need for human factors studies differ greatly from what traditional pharma is used to.
This two-part article addresses the issues that pharma companies face when moving into the digital health space. This first installment explores the development process, agency guidance, and need for quality oversight for software incorporated into medical device and combination products.
What Is A Combination Product?
A combination product is composed of a drug and a device, a biological product and a device, a drug and a biological product, or a drug, device, and a biological product. They are regulated by the FDA’s Office of Combination Products (OCP), which takes the lead in deciding which FDA center will have primary jurisdiction for a product’s premarket review and regulation, based on the product’s primary mode of action (PMOA).
However, when software or firmware components are part of the product, numerous other development considerations, agencies, and regulations also come into play. For example, if a medical device or combination product transmits data wirelessly to a cloud for physicians or others to view or evaluate, the Federal Communications Commission (FCC) has regulations that must be considered.
At the Xavier Health Combination Products Summit, Suttons Creek managing partner Steven Badelt provided a review of the evolving digital health regulatory landscape. Badelt has 20 years’ experience in neuroscience, systems engineering, and medical device design and manufacturing, and has consulted with numerous pharma companies as they moved into the medical device and combination product spaces, especially with products containing digital health components.
“It is not necessarily an easy transition for pharma, going from more of a focus on a discovery and manufacturing culture to a design culture,” he said. “I see challenges in the next few years for pharma as it tries to wrap its head around how it is going to put software into its quality systems and bring it into the fold for their companies.”
Designing A Combination Product’s Development Process
The Venn diagram below shows how the development processes for drugs, devices, and software overlap with respect to the various types of combination products, including those that contain software.
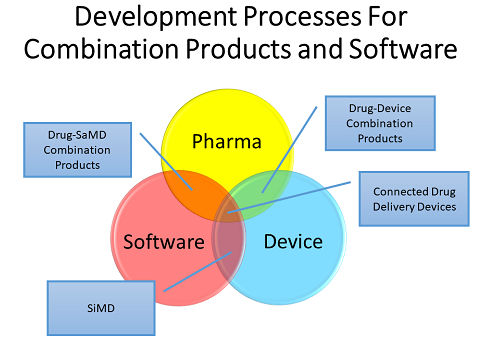
Software can be incorporated into combination products and medical devices in a variety of ways, each with its own unique set of challenges. These include drug-device combination products with software as a medical device (SaMD) — which includes software medical device apps supporting a drug product – and software in a medical device (SiMD).
Other possibilities include an injection system on a body or an insulin pen — a system that connects to a cell phone and transmits data to the cloud, termed a “connected delivery device.” Software can be embedded as firmware or as part of a medical device that has a hardware component.
Most firms develop a combination product by combining the drug and device processes in a streamlined approach by using the original CMC quality system and adding on the medical device components — for example, as outlined in CFR 211 820.30, Quality System Regulation — to make sure the company is compliant for device manufacturing. The combination product life cycle process differs from those used in pharma and medical device manufacturing (see figure below).
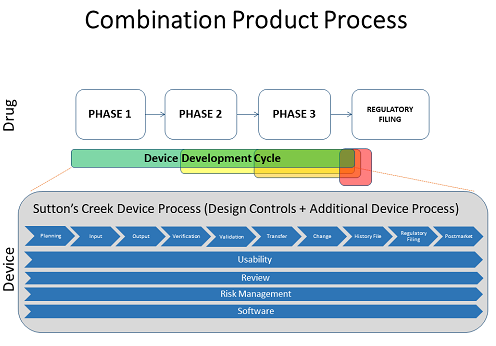
The phases included in the design control process from CFR 820.30, such as planning, input, output, verification, validation, and so on — are things the industry is fairly familiar with. The bars below that – usability, review, and risk management — are things that should be included across the entire development process.
The process model is commonly known as a “waterfall” method, with a series of steps one after another in a long flow of activities that end up at the final product — a model very different than that used in software development.
Software Guidances Are Numerous And Varied
Two of the primary guidances for combination product development are 21 CFR 820.30 and ISO 13485. Software development practices are overlaid on top of those. While these are not entirely different, there are specific elements of software design and development that should be used in conjunction with those guidances, including ISO 14971 with respect to risk management.
The following is a list of guidance documents and agencies that are important in the software space.

The FDA 2015 Mobile Medical Applications guidance is a key document to look at, as is the FDA guidance for premarket submissions, to understand the agency’s thinking on deliverables for software. Also useful for software life cycle guidance is the international standard IEC 62304, which specifies life cycle requirements for the development of medical software and software within medical devices.
Cybersecurity and cybersecurity risk management must also be considered, with protections put in place to ensure that patient data, and potentially company data, is secure from attack.
Guidance on how to incorporate software development practices into a pharma quality system comes from the International Medical Device Regulators Forum (IMDRF). Specifically, guidance documents N10, N12, N23, N35, and N41 have very good descriptions of how to roll these things in, including the implications.
The Association for the Advancement of Medical Instrumentation (AAMI) TIR45 is relevant to agile software development practices.
While software development does follow risk-based approaches, it differs from the approach used in medical device development. With software, the level of risk to the patient determines what level of documentation and what level of detail are needed in development practices.
“While we certainly have to follow risk management, FDA and IEC are more specific in terms of which deliverables and which execution you need to be performing and at what level for software,” Badelt explained. The primary guidances for the approach and deliverables are FDA Premarket Submissions for Software Contained in Medical Devices – Level of Concern and IEC 62304.
Each has a similar three-tier system for classifying risk. The higher the risk, the more documentation and code review and work must be done to develop the software and be sure that you have covered things appropriately for the patient.
In terms of what is or is not considered a medical device, the FDA’s Mobile Medical Applications (MMA) Guidance – Enforcement Discretion outlines the enforcement discretion areas, along with the 21st Century Cures Act update, in terms of what the agency considers a medical device.
If a firm does not think a product is a medical device or is under enforcement discretion, “you need to be going through a risk-based approach within your company and have that process defined so that you can provide evidence to the agency that you did the analysis and show how you arrived at your conclusion,” Badelt advised.
Other regulators that may need to be considered include the Federal Trade Commission (FTC), the European Union, and the FCC. For example, if the software is developed as an app and is available on Apple’s iTunes store or Google Play, any removal or changes are enforced by the FTC. Regarding patient data, the European General Data Protection Regulation (GDPR) governs what can be sent to patients, what data can be monitored, and who it can be shared with.
Companies considering the use of wireless technology as part of a medical device will need to understand the appropriate FCC regulations. If a device transmits a signal wirelessly into the cloud and the doctor needs to know immediately, for example, that the patient has not gotten his or her dose, the regulatory requirement would be to demonstrate that the wireless signal reliably gets through.
Also important for a software app is the required end user license agreement (EULA), which will need to be developed with both legal and regulatory input. The EULA “is going to specify in many cases what the intended purpose of the device might be, or it may have flavors thereof. You will need to be very careful that what it says aligns and how it aligns with your regulatory filing,” Badelt added.
Software Development Requires Quality Oversight
Software development includes a number of activities that will be familiar to medical device companies, and many that will not (see figure below). Design and development documents or processes that a company does not have will need to be developed within your company, or you will have to have quality system upgrades in place that can cover these types of things when the development is performed by an external vendor.

Familiar processes include requirements analysis, verification, and design, which are detailed in IEC guidance 62304. Other activities will be similar to those already performed but may have different names. For example, “detailed design documentation” is similar to “specifications.”
One unfamiliar term may be “COTS,” or “commercial off the shelf software.” How is it dispositioned and when it can be used requires additional activities on top of those for software developed in-house.
One area that Badelt feels will need attention is software quality audits and how they are performed at contract or partner companies. “The vast majority of the companies that you might hire for software development do not have a quality system in place,” Badelt said. “You will have to make very clear decisions on how you will be managing those as a pharmaceutical company, and whether you can put a quality wrapper around them or whether you are going to find a more expensive house that may have their own quality system. Those are quite limited in number right now.”
Part 2 of this two-part article will focus on the importance of cybersecurity and software maintenance, how time scales and methods of execution differ from those in traditional pharma, how agile software development works, and how to avoid potential pitfalls.
About The Author:
 Jerry Chapman is a GMP consultant with nearly 40 years of experience in the pharmaceutical industry. His experience includes numerous positions in development, manufacturing, and quality at the plant, site, and corporate levels. He designed and implemented a comprehensive “GMP Intelligence” process at Eli Lilly and again as a consultant at a top-five animal health firm. Chapman served as senior editor at International Pharmaceutical Quality for six years and now consults on GMP intelligence and quality knowledge management and is a freelance writer. You can contact him via email, visit his website, or connect with him on LinkedIn.
Jerry Chapman is a GMP consultant with nearly 40 years of experience in the pharmaceutical industry. His experience includes numerous positions in development, manufacturing, and quality at the plant, site, and corporate levels. He designed and implemented a comprehensive “GMP Intelligence” process at Eli Lilly and again as a consultant at a top-five animal health firm. Chapman served as senior editor at International Pharmaceutical Quality for six years and now consults on GMP intelligence and quality knowledge management and is a freelance writer. You can contact him via email, visit his website, or connect with him on LinkedIn.
Image credit: Proteus Digital Health