
Pharma 4.0: A New Framework & Process For Digital Quality Management
By Bikash Chatterjee, CEO, Pharmatech Associates
 A little over 10 years ago I wrote an article urging our industry and the FDA to truly embrace the 2004 FDA guidance1 and the shift to scientific understanding as a basis to ensure product quality, safety, and effectiveness. Up until this landmark guidance, the industry had operated under the assumption that inspection and testing could ensure the suitability of products for the marketplace. The quality struggles related to the rapid global expansion of the supply chain revealed the foresight of this fundamental change, which opened a way forward to remaining competitive while ensuring safe and efficacious products.
A little over 10 years ago I wrote an article urging our industry and the FDA to truly embrace the 2004 FDA guidance1 and the shift to scientific understanding as a basis to ensure product quality, safety, and effectiveness. Up until this landmark guidance, the industry had operated under the assumption that inspection and testing could ensure the suitability of products for the marketplace. The quality struggles related to the rapid global expansion of the supply chain revealed the foresight of this fundamental change, which opened a way forward to remaining competitive while ensuring safe and efficacious products.
A road map for transformation slowly began to emerge as the regulatory authorities in the U.S. and Europe updated their guidance and directives, seizing upon mutually agreed industry best practices to guide regulatory compliance. Harmonization at the regulatory level spurred industry’s recognition of the need for transformation, not just at the execution level of the drug development process, but also upstream at the discovery level.
Our industry lagged behind other industries in terms of productivity and efficiency. We were hampered by the false notion that regulatory compliance and innovation cannot coexist. For many drug developers and manufacturers, innovation was stifled by an outdated documentation and compliance philosophy that limited our ability to seize upon competitive market opportunities, to effectively manage our cost of goods sold (COGS), and to optimize the ever-expanding complexity of our global supply chain. There was no incentive to improve how we manage and measure product quality. More importantly, we did not think of quality as a driver for competitive advantage — it was just a necessary component of compliance.
Quality And The Transformation Of Drug Development
Today’s R&D and compliance environment bears little resemblance to the framework of the last 50 years. Several events triggered this evolution. First, in the U.S., the issuance of the 2011 FDA guidance on process validation forced the industry to look closely at quality and adopt the principles of quality by design (QbD) captured in ICH (International Conference on Harmonisation of Technical Requirements for Pharmaceuticals for Human Use) Q8. At the same time, the FDA redefined process validation as a life cycle concept rather than the final step in product development before commercialization. We cannot overstate the impact of this change. No longer could we rely simply on satisfying our product specifications during validation: QbD forced us to define what was important from a process and material characterization perspective before trying to argue process predictability.
Identify the differences between the old GMP mindset and the new quality systems mindset in the course:
Quality by Design (QbD): Making Sense of the ICH Q8, Q9, Q10 Puzzle
If scientific understanding were to be the basis for defining quality and suitability, then the framework for administering quality had to change as well. To temper the impact from this radical shift in thinking, the FDA supported integrating quality risk management (QRM) within product and process design. This is the underlying principle behind industry best-practice guidance such as ICH Q10. Until Q10 was released, the industry sought to enforce a framework that separated the quality organization from the development organization, positioning quality as the ultimate decision-making authority. However, Q10 advocated that quality and process development be integrated — not separated — if an organization were to achieve both performance and control. This translates to a quality organization grounded in scientific understanding and management of risk, with a solid foundation in statistical analysis.
The best illustration of the new quality mindset is the administration of an organization’s stage III process validation program called continued process verification (CPV). CPV provides a framework for making process adjustments for new and legacy products without repeating stage II process qualification. For the quality organization, this requires establishing predicate requirements around process understanding, expected process behavior, and product variation. It also requires creating a mechanism for adjusting the process for commercial product.
This would be impossible without a keen understanding of what does and does not matter in terms of expected process and product performance. Quality professionals have to understand how phase-appropriate data relates to making decisions about quality. This distinction illustrates the central difference between ensuring product quality through scientific understanding vs. simply ensuring QMS adherence, traceability, and specification compliance.
A New World Of Data And Analytics
We have seen a renaissance in treatment therapies over the last decade with new and significant drug modalities emerging. Antibody-drug conjugates (ADCs), chimeric antigen response T-cell therapy (CAR-T), next generation gene sequencing (NGS), clustered regularly interspaced short palindromic repeats (CRISPR/Cas9 targeted genome editing), and the field of nanomedicines promise to provide us with the disease mechanism insight and drug delivery solutions to achieve unprecedented progress against major disease states. The evolution of these drug modalities and diagnostic tools has elevated our approach to data analysis, moving data analytics from the back room to the boardroom.
Industry regulators have marked this step with new analytics frameworks. The FDA issued its guidance on quality metrics for the drug industry in 2016, recognizing that an organization’s quality management system (QMS) is comprised of discrete, high-performing processes that require a clearly articulated governance structure. Buoyed by a decade of operational excellence initiatives, industry is aware of the benefits of measuring performance and the use of key performance indicators (KPIs), even if it has been slow to respond to the guidance.
Today, many drug companies have shifted completely away from vertical product development and manufacturing to concentrate on how best to utilize contract service providers (CSPs) as their surrogate for internal staff. The need to acquire and share data internally with the drug sponsor is central to the outsourced strategy, but some manufacturers are evaluating whether it is possible to share data between service providers to drive performance. From a starting point of industry risk-aversion, the regulators have empowered manufacturers to keep evolving to combine industrial processes with new advanced analytics capability. The approach known as Pharma 4.0 represents a strategy for the future, derived from an intelligent and fully connected network across the entire value chain.
Digital Quality Management And Pharma 4.0
Pharma 4.0 is a term for the industry-specific global vision and initiative based on Industry 4.0. The idea behind Industry 4.0 is to develop the infrastructure and create standards that allow us to connect data, physical equipment, and human resources in the context of an overarching cyber-physical system. Of course, Pharma 4.0 promises to improve quality, productivity, and lead times through interconnectivity and automation, but the most dramatic potential is the ability to gather data beyond the four walls of the factory through the use of Internet of Things (IoT) and Big Data analytics. Cloud-based data management and advanced analytics are fundamental components of Pharma 4.0, and full system integration will lead to enormous data volumes that have to be analyzed, stored, shared, and protected across the global supply chain and the development value chain. The basic tenets of Pharma 4.0 are captured in Figure 1.
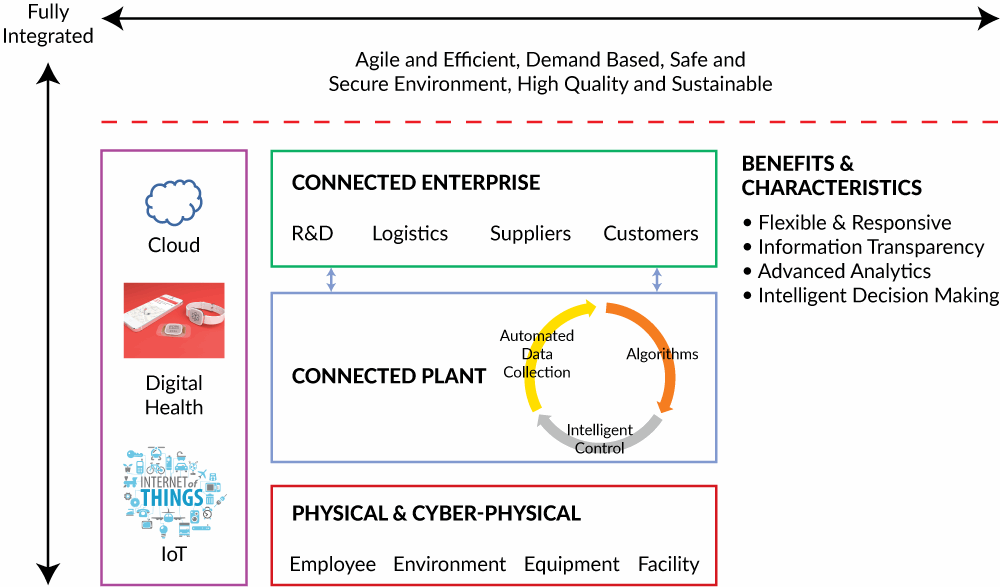
Figure 1: Components of Pharma 4.0
The Pharma 4.0 vision is ambitious. At the physical level, intelligent devices gather data from every facet of the organization. On the shop floor and in the lab, cyber-physical systems will be capable of making informed decisions, adjusting as required to perform tasks autonomously. Across the connected plant, analytics will evaluate all input and output data, measuring and adjusting operations on an ongoing basis. The connected enterprise will gather and provide data from every step within the overall value chain. From drug discovery through post-market monitoring, the data will be available for analysis.
Digital health promises to provide the missing piece in long-term drug performance monitoring via pharmacovigilance; a shift toward information transparency assumes the competitive pharmaceutical company of the future will be able to break through the silos that have long inhibited collaboration and efficient data communication within classical pharmaceutical organizations. These principles are not new. The philosophy of architecting a work environment and systems based on the free exchange of ideas and information across nontraditional areas of expertise (molecule design, immunology), has worked brilliantly to catalyze innovation in post-doctoral research institutes such as the Gladstone Institutes in San Francisco. What is new is the shift to a structured framework that starts with data gathering and analysis and moves through deployment. The biggest promoters of Pharma 4.0 are manufacturing execution system (MES) service providers such as Werum and Rockwell Automation, largely because of the critical role in information connectivity that MES systems can play within the four levels of ISA 95.2
Predicate Requirements
Automation and information management are not a panacea: The core challenges of data quality and integrity, security, and analytical expertise that face us today will persist. To exploit the benefits of a fully connected ecosystem, we must consider certain characteristics when designing and offering Pharma 4.0 solutions (outlined in Figure 2).
Today’s issues must lead in the design of both the technology and architecture of information integration. Data integrity has become a critical issue in the emerging markets and it will be a core element in any Pharma 4.0 rollout plan. When patient engagement and clinical data are involved, then global privacy compliance requirements must also move to the forefront of any solutions contemplated.
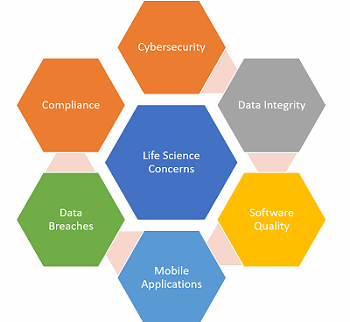
Figure 2: Life science issues
Cybersecurity And Data Compliance
Commensurate with the risk, new emphasis and investment in cybersecurity must grow to maintain the integrity of future interconnected systems. As pharma continues to expand its dependence on data as a foundation for drug development and clinical trial management, having a nimble, robust cybersecurity program will be a high priority. In current practice, cybersecurity is not the primary consideration when designing and implementing an information management system. Look to future cybersecurity approaches to harmonize and move to the head of many organizations’ strategic plans.
Conclusion
Since the 2004 FDA “compliance through science” guidance was first issued, drug quality and business performance have improved. The bold changes undertaken 10 years ago will allow regulators and industry to take advantage of the incredible innovations we are poised to see in healthcare. We are moving toward a framework and process that will allow us to acquire and handle data, making Pharma 4.0 a reality. But, the devil is in the details. If solutions providers sharpen their focus on the issues plaguing the industry today, the industry will transition to new self-aware, predictive technology components and architectures with minimal disruption. If, however, we relegate the core issues of data integrity and cybersecurity to the background, the benefits of a new era of safety and efficiency promised by Pharma 4.0 will likely remain out of reach.
References:
- FDA Guidance for Industry: Pharmaceutical CGMPS for the 21st Century -- A Risk Based Approach, https://www.fda.gov/drugs/developmentapprovalprocess/manufacturing/questionsandanswersoncurrentgoodmanufacturingpracticescgmpfordrugs/ucm137175.htm
- ISA95: Enterprise-Control System Integration, https://www.isa.org/belgium/standards-publications/ISA95/
About The Author:
 Bikash Chatterjee is president and chief science officer for Pharmatech Associates. He has over 30 years’ experience in the design and development of pharmaceutical, biotech, medical device, and IVD products. His work has guided the successful approval and commercialization of over a dozen new products in the U.S. and Europe.
Bikash Chatterjee is president and chief science officer for Pharmatech Associates. He has over 30 years’ experience in the design and development of pharmaceutical, biotech, medical device, and IVD products. His work has guided the successful approval and commercialization of over a dozen new products in the U.S. and Europe.
Mr. Chatterjee is a member of the USP National Advisory Board, and is the past-chairman of the Golden Gate Chapter of the American Society of Quality. He is the author of Applying Lean Six Sigma in the Pharmaceutical Industry and is a keynote speaker at international conferences. Mr. Chatterjee holds a B.A. in biochemistry and a B.S. in chemical engineering from the University of California at San Diego.