
Outsourcing And Phase-Appropriate GMP For Clinical Manufacturing Needs
By Ajay Pazhayattil, Michelle Gischewski, and Salman Pathan

Appropriate manufacturing and timely delivery of investigational products are essential for the successful execution of clinical trials, and, ultimately, for the success of a drug development program. Challenges are common in clinical manufacturing, especially in CDMOs. This article provides a high-level overview of these common challenges, along with best practices to address them.
CDMO Onboarding
Establishing confidential disclosure agreements (CDAs) and manufacturing services agreements (MSAs) is critical. During the initial negotiations, companies must establish rigorous safeguards to protect their proprietary data and trade secrets. Liability issues can pose significant challenges. Determining the party responsible for potential breaches, mishaps, or losses can be complex and contentious. Hence, the negotiation phase can be prone to delays. When companies cannot reach a consensus on the terms or when bureaucratic and legal hurdles impede progress, it can hinder the timely execution of the clinical project. A protracted negotiation process also can introduce uncertainty and risk into the partnership itself, potentially causing missed initial deadlines.
A thorough assessment is pivotal in onboarding compounds for formulation development and clinical manufacturing. This activity involves amalgamating all available product/similar product information and toxicology data. The outcome is the decision regarding whether to proceed or not. The compound banding questionnaire (CBQ), an information checklist, is diligently completed to commence this process. The purpose of the CBQ (refer to Table 1) is to aid in the calculation of the occupational exposure limit (OEL) and acceptable daily exposure (ADE) values. The data collected serves as a critical starting point for toxicologists' assessments, allows for identifying and initiating effective risk mitigation and engineering control strategies to ensure employee safety, and helps determine the proper cleaning methods for the compound. The safety assessment report (SAR), which is the outcome report, outlines the assigned ADE value, which is instrumental in establishing the cleaning limits when multi-use equipment is used.
Table 1: An Example of Compound Banding Questionnaire (CBQ)

Indicate available information and attach supporting data reports where applicable.
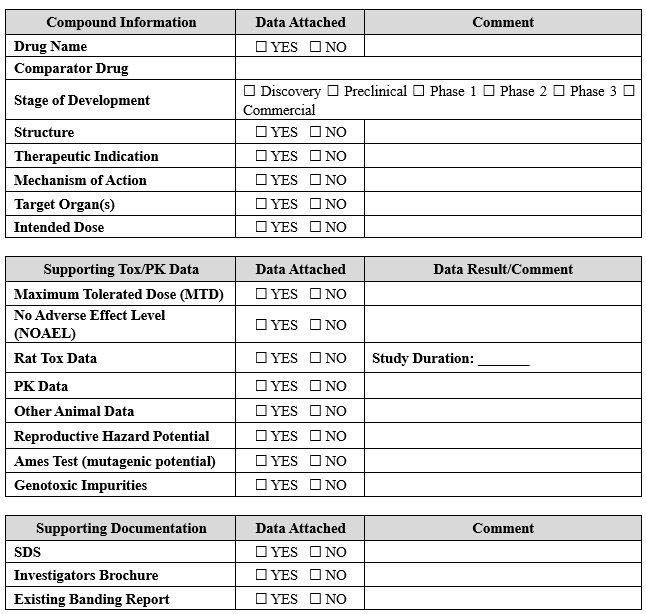
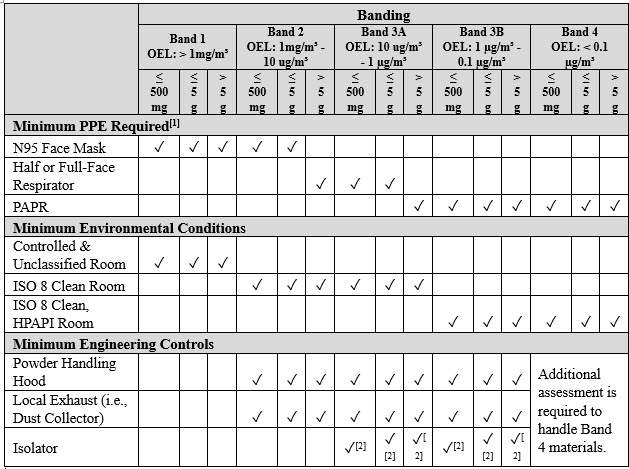
After completing the CBQ and generating the SAR, a comprehensive evaluation is conducted to determine the requisite engineering controls, PPE, process controls, and facility location to effectively manage the compound as per the designated OEL value. The specific handling protocols are contingent upon the safety banding category and the quantity of material to be managed, with the final detailed procedures clearly outlined in the batch record, standard operating procedure, or work instructions. Determining the minimum PPE, environmental conditions, and engineering controls for handling is intimately linked with the safety banding and the quantity to be handled (refer to Table 2). These parameters are meticulously assessed to ensure the highest levels of safety. Before the commencement of any project, a comprehensive assessment, such as the process hazard review, is undertaken to delineate the project's process steps, identify potential hazards, and establish effective risk mitigation strategies. Integrating risk assessment procedures into onboarding is fundamental to ensuring employee safety and effectively managing compounds in clinical manufacturing. This proactive approach minimizes risks and enables the smooth execution of clinical trial batches in a controlled, efficient, and secure fashion.
Table 2: An Example of Minimum PPE, Environmental Conditions and Engineering Controls for Handling Based on Safety Banding and Quantity
Phase-Appropriate GMP Manufacturing
Phase-appropriate GMP (refer to Table 3) entails the application of well-defined GMP criteria suitable for the specific stage of clinical development, where the manufactured and tested material will be utilized. For instance, during Phase 1 clinical trials, the primary focus is assessing the drug's safety. In contrast, in later-phase clinical trials and post-marketing phases, the emphasis shifts toward expanding the utilization and improved understanding of the product and its clinical application.
Table 3: An Example of a Phase-Appropriate Strategy1
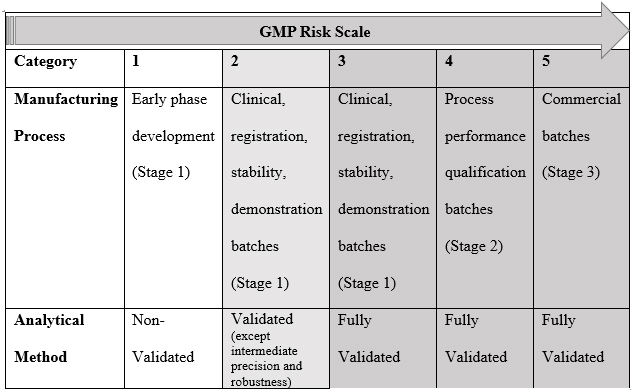
As a drug advances through the clinical development stages and approaches market approval, adherence to the GMP standards becomes increasingly crucial in ensuring the final product's quality and consistency (minimizing variability). Investigational medicinal products (IMPs) must adhere to GMPs, and it is the manufacturer's responsibility to outline and justify the practices they follow for manufacturing materials for each clinical phase while considering any market-specific requirements. In certain circumstances, the sponsor/client may specify the need to follow certain additional requirements (for internal policy or product reasons); however, they should not be confused with the mandatory requirements. Creating organizational clarity in this respect is pivotal to clinical manufacturing success.
It must be noted that clinical manufacturing activities also include producing placebo or control drugs for use in the trials. To ensure the drug's safety, efficacy, and compliance with regulatory standards,2 clinical packaging, labeling, release, and shipping must also meet the rigorous GMP standards. This necessitates the inclusion of appropriate labels providing essential information about the drug, including its name, strength, dosage form, and any necessary warnings or precautions. Clinical trial packaging requirements vary across phases, with Phase 1 favoring small, adjustable packaging, Phase 2 needing larger containers, and Phase 3 employing multi-dose options. At the same time, special considerations arise for pediatric trials, placebo controls, cold chain products, adaptive trials, and biosimilar trials. Participants may require user-friendly, clearly labeled packaging and organized compartments to simplify the dosing schedule. Flexibility in packaging is essential to accommodate changes in trial protocols, such as adjustments in dosing regimens or additional study arms, ensuring seamless adaptation during the trial.
As a manufacturer, it is important to identify and understand the regulatory, patient, and client commitments and risks before commencing a clinical development and manufacturing project. The expectations and goals must be established and communicated to all stakeholders.
Key Considerations In Clinical Manufacturing
Various challenges can arise in clinical manufacturing that affect the manufacturing and supply timeline. To maintain a smooth process, it's crucial to implement strategies to mitigate these issues. Delays in supplier manufacturing can result in insufficient essential raw materials or active ingredients. Custom containment and engineering solutions can cause design and construction delays in containment and special manufacturing needs. To counter this, proactive measures such as forecasting, working closely with drug substance facilities, identifying key suppliers, diversifying suppliers, and maintaining adequate inventory can help.
Shipping delays, losses, or damage to materials also can disrupt production timelines. Minimize these risks by choosing reliable shipping carriers and closely monitoring shipment progress to ensure on-time delivery.
Quality issues with raw materials, like impurities or variations in potency, can compromise the safety and efficacy of the final product. Rigorous quality control measures and early development of test methods are essential to address these concerns effectively.
Unvalidated methods and processes can lead to unforeseen issues during clinical manufacturing stages. Allocate additional time to identify and resolve potential manufacturing process problems to prevent delays.
Suppose substantial modifications are introduced to the investigational or comparator product along clinical development. In that case, it is crucial to have the results of any supplementary studies on the reformulated product (e.g., stability, dissolution rate, bioavailability) available before implementing the new formulation for clinical trial materials. This is necessary to evaluate whether these changes would have a notable impact on the pharmacokinetic profile of the product.
The clinical trial material, which encompasses active comparators and, when relevant, placebos, should be appropriately characterized based on the product's development stage.
The packaging and/or kits should be designed to prevent contamination and unacceptable deterioration during transport and storage during clinical supply. Cold chain packaging solutions ensure products maintain a consistent temperature during transit, whether domestically or internationally. Phase change materials (PCMs)3 have revolutionized clinical trial material packaging with their ability to store and release heat while maintaining temperature. Clinical material is often costly or only available in small quantities, and product loss most likely leads to extensive delays in re-manufacturing. A data-driven approach involving stakeholder input is crucial to create an optimal PCM-based solution. This identifies essential product parameters, such as temperature control, logistics, and transit routes, and employs risk management, technical reviews, and traceability.
Clinical sites, warehouses, and distribution centers must be well-equipped with cold storage, refrigeration, and monitoring devices. These facilities play a vital role in properly storing prescription drugs and temperature-sensitive clinical supplies. Consistency is key, as cold chain packaging configurations must meet quality standards and operational expectations, even in the face of clinical supply logistical uncertainties and occasional human errors.
Customs problems during distribution, such as import/export complications across global sites or while handling hazardous materials, can create hurdles. Ensure customs clearance requirements are followed and procedures are established to manage issues promptly. In this case, having an assigned broker to facilitate cross-border shipments can be advantageous.
Trial materials should be coded and labeled to safeguard the blinding, where applicable. In blinded trials, the coding system for the investigational product must allow for swift product identification in the event of a medical emergency. Still, it should not enable undetectable breaches of blinding. Furthermore, the labeling must adhere to relevant regulatory standards. The specifications for the product should encompass acceptable storage temperatures, storage conditions (e.g., protection from light), storage durations, reconstitution fluids and procedures, and any required product infusion devices.4 Organizations can enhance operational efficiency by harnessing existing serialization and track & trace infrastructure.
Conclusion
The thorough safety assessments during the onboarding phase are significant and involve meticulous analysis of toxicology data to establish effective risk mitigation and engineering control strategies, thereby ensuring employee safety and proper cleaning methods for the compounds used in clinical manufacturing. In addition to safety assessments, adopting phase-appropriate GMP criteria for the specific stage of drug development is critical. Proactive measures are needed to address potential challenges such as delays in supplier issues, quality control concerns, and shipping problems. The clinical trial regulations necessitate precise labeling, proper storage, and adequate packaging solutions to maintain the quality and safety of clinical trial materials. In a competitive environment, purpose-fit strategies are essential to navigate the intricacies of clinical trial material manufacturing effectively.
References
- https://www.americanpharmaceuticalreview.com/Featured-Articles/518306-A-Roadmap-for-Implementing-QRM-Principles-in-Drug-Substance-Manufacturing/
- https://www.raps.org/products/fundamentals-of-pharmaceutical-and-biologics-regulations-a-global-perspective
- https://www.pharmamanufacturing.com/development/qbd/article/11288521/taking-a-qbd-approach-to-packaging-development
- https://database.ich.org/sites/default/files/E6_R2_Addendum.pdf
About The Authors:
 Ajay Pazhayattil is a management consultant and an industrial pharmacist with pharmaceutical operational experience in the industry's sterile, solid oral, and API sectors. He has been in leadership roles with brand, generic, and CDMO organizations before establishing cGMPWorld. Pazhayattil recently contributed to the RAPS book Fundamentals of Pharmaceutical and Biologics Regulations: A Global Perspective, Chapter 12: Coordinating Drug Supply for Clinical and Nonclinical Development. He can be contacted through LinkedIn.
Ajay Pazhayattil is a management consultant and an industrial pharmacist with pharmaceutical operational experience in the industry's sterile, solid oral, and API sectors. He has been in leadership roles with brand, generic, and CDMO organizations before establishing cGMPWorld. Pazhayattil recently contributed to the RAPS book Fundamentals of Pharmaceutical and Biologics Regulations: A Global Perspective, Chapter 12: Coordinating Drug Supply for Clinical and Nonclinical Development. He can be contacted through LinkedIn.
 Michelle Gischewski is a consulting leader in the pharmaceutical industry. She has a solid background in pharmaceutical science, with expertise with critical new product development projects and clinical manufacturing. She possesses technical knowledge in R&D formulation development, process development, technology transfer, scale-up, and qualification of solid and liquid dosage forms. As the head of drug product formulation consulting, she is responsible for optimizing novel formulations and streamlining processes to ensure robustness. She can be contacted through LinkedIn.
Michelle Gischewski is a consulting leader in the pharmaceutical industry. She has a solid background in pharmaceutical science, with expertise with critical new product development projects and clinical manufacturing. She possesses technical knowledge in R&D formulation development, process development, technology transfer, scale-up, and qualification of solid and liquid dosage forms. As the head of drug product formulation consulting, she is responsible for optimizing novel formulations and streamlining processes to ensure robustness. She can be contacted through LinkedIn.
 Salman Pathan is a senior global clinical supply expert, a serial entrepreneur, and an investor. He has spearheaded expansion on a global scale. His services are focused on clinical supply and specialty medicine sourcing, distribution, pharma licensing, partnering, and mergers and acquisitions. His operational presence spans across the United States, Canada, Europe, and Asia. He can be contacted through LinkedIn.
Salman Pathan is a senior global clinical supply expert, a serial entrepreneur, and an investor. He has spearheaded expansion on a global scale. His services are focused on clinical supply and specialty medicine sourcing, distribution, pharma licensing, partnering, and mergers and acquisitions. His operational presence spans across the United States, Canada, Europe, and Asia. He can be contacted through LinkedIn.