
New AAV Reference Standards To Aid CQA Assessment
Anthony J. Blaszczyk, United States Pharmacopeia

The complexity of AAV products makes characterization difficult. This challenge is further compounded by the lack of harmonization among analytical methods, as well as the lack of reference standards to support analytical testing.1,2 For example, there are more than a dozen analytical methods currently being used to assess AAV empty/full content, but companies lack the bandwidth and material to test each method, so it ultimately leads to vast differences in reportable results.3-5
However, publicly available AAV reference materials are not sufficient for most analytical testing laboratories. They lack thorough characterization, such as being assessed by, at most, only one empty/full method, and they have insufficient volume and concentration for many common physicochemical tests.
As part of a suite of reference standards — including for starting materials, which is described in an earlier discussion — USP is developing analytical reference standards and reference material to support the characterization of AAV viral vectors, as well as the assessment of common impurities, such as residual plasmid and genomic DNA. To best help users, USP is using available resources to thoroughly characterize all our AAV products, which often involves using multiple testing methods and multiple labs to assess a single critical quality attribute (CQA).
AAV8 Reference Standards — Empty And Full Capsids
Knowing the number of full AAV capsids is critical for both safety and efficacy of the final drug product, as empty and partial capsids are impurities of concern, according to FDA guidance for human gene therapy.6 Also, empty AAV capsids do not deliver the gene of interest to patients but may cause a harmful immune response. Although being of utmost importance, accurate quantitation of full and empty capsids remains a major challenge in the field, as there remains a lack of method harmonization and physical standards.3,5 There are more than 10 analytical methods currently being used by different laboratories to quantify the percentage of full capsids, highlighting the diversity in testing. The variation between techniques makes alignment difficult, especially when a physical reference standard is not commercially available. AAV reference materials that are currently on the market generally lack thorough characterization and only quantify the percentage of empty and full capsids by a single method.
To aid AAV analysts in empty/full characterization, USP is developing two AAV reference standards, AAV8 (Empty Capsids) and AAV8 (Full Capsids). These reference standards will be well characterized in a multi-lab study that utilizes five unique methods – analytical ultra centrifugation (AUC), size exclusion chromatography with multiple angle light scattering (SEC-MALS), mass photometry, UV-vis, and charge detection mass spectrometry (CD-MS) – to quantify the percentage of empty and full capsids in each standard. Furthermore, these standards will be offered in high titer concentration and relatively high volume, so one unit will be sufficient for nearly every physicochemical test, even ones that have higher sample requirements, such as AUC. The characterization via multiple analytical techniques will be extremely advantageous to all laboratories, regardless of their primary characterization method, as it will either allow for a direct comparison or allow assessment against multiple orthogonal methods.
In addition to the array of empty/full characterization tests being performed on USP’s AAV reference standards, the following critical quality attributes will be analyzed: appearance, capsid purity, purity (aggregation), capsid titer, genomic titer, and genome integrity. These additional characterization tests will provide users with a more thorough understanding of the USP product, allowing them to expand the scope of use for this AAV standard.
AAV Capsid Titer Reference Material
AAV capsid quantitation is a critical quality attribute that measures the number of specific capsid particles in each sample. Various titer methods can be used for capsid measurements, but serotype-specific ELISA kits are the most common for measuring particle concentration. These ELISA assays, however, are known to have drawbacks, including lack of robustness and long reaction times, and they can lack standardization to an orthogonal method.7 In each AAV ELISA assay, an AAV standard is used to generate the calibration curve, which is subsequently used to quantify capsid content. Other immunoassay titer methods have begun emerging as ELISA alternatives, but they too rely on an AAV calibrant for quantitation. Because the AAV standards used in the various titer assays are not standardized against an orthogonal method, they are a potential source of much inter-assay variability. USP is addressing this issue by developing serotype-specific AAV capsid standards that could be used primarily for standards in capsid titer assays. These standards will be quantified using an orthogonal method (SEC-MALS), allowing for a truly independent measurement of the actual capsid content. Additionally, these materials will all have a five- to seven-year target shelf life, which ensures users that they will be receiving the same material from the same lot for an extended period of time — further increasing the reliability of the USP product. These materials will be prime candidates to either be used as positive controls or calibrants in immunoassays that require calibration curves. These USP capsid standards can help users in a number of ways, including ensuring the immunoassay’s accuracy, serving as a standard for bridging studies when the calibrant lot is changed, and comparing between kits, either by use as a calibrant or as a normalization factor.
USP’s AAV capsid titer reference materials comprise serotype-specific AAV capsids (empty) in liquid form, as lyophilization can alter the three-dimensional structure of proteins. This physical quality is particularly critical for immunoassays, as measurements are dependent on proper antibody binding, which can be influenced by the three-dimensional protein structure. SEC-MALS, which is an orthogonal method that first separates by size and quantifies capsid particles using UV and light scattering data, will be used to determine the capsid titer value of our reference materials. These materials will also be valuable in qualifying/validating capsid titer assays, as having a well-characterized control is advantageous in these activities.
Fig. 1
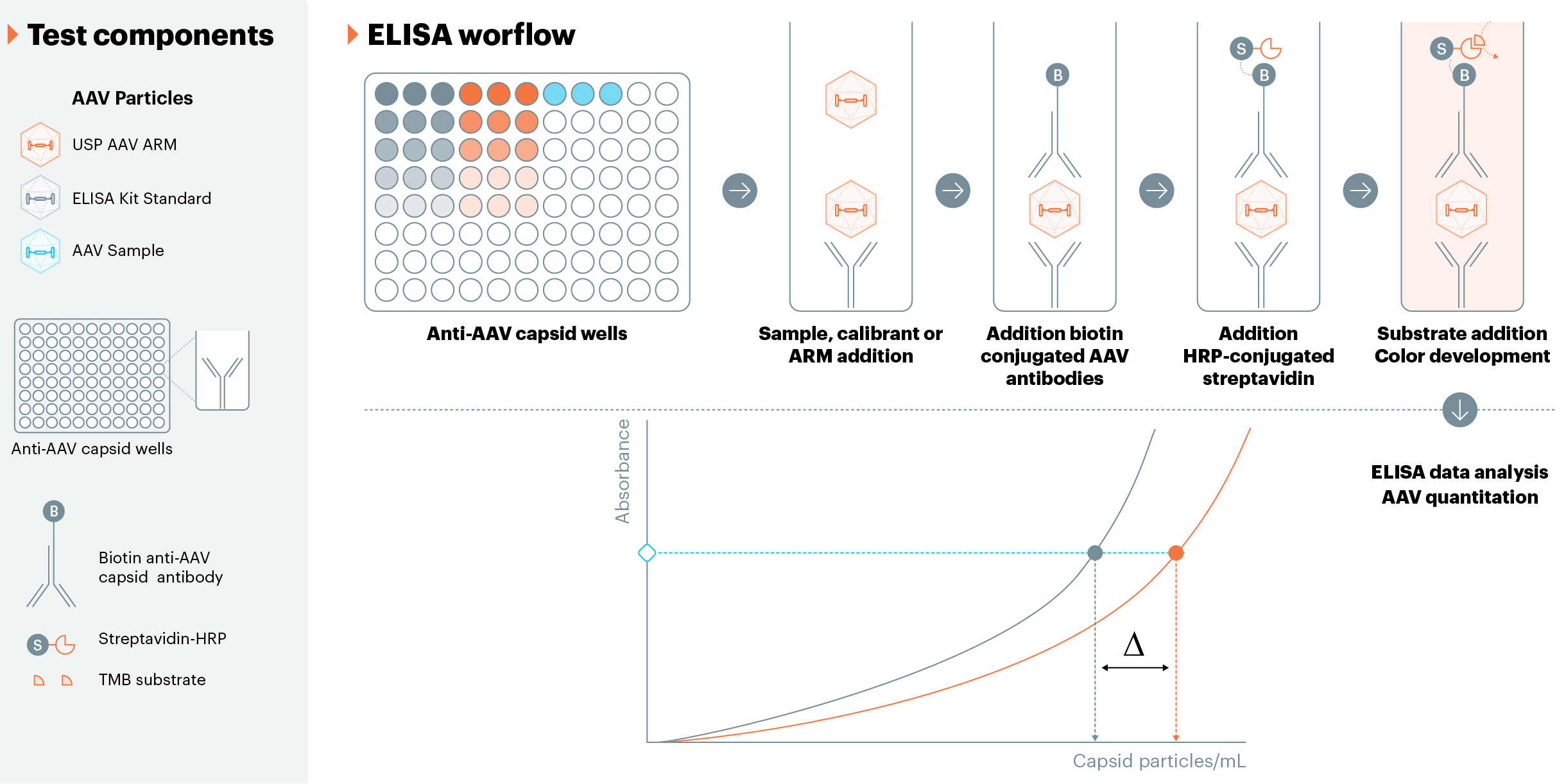
gDNA (HEK293 And Sf9) Reference Material
During the AAV production cycle, it is critical to remove genomic DNA (gDNA), as residual host cell gDNA, which can harbor oncogenic or viral sequences, poses a significant safety risk to patients.5 In fact, to emphasize the critical importance of gDNA clearance in all drug products, multiple regulatory agencies, including the FDA, European Medicines Agency (EMA), and WHO, have set a specification limit for the maximum allowable amount of gDNA (10 ng/dose, ≤ 200 bp in length).8-10 Quantitative PCR (qPCR) is the most widely used analytical method to quantify residual gDNA. Although qPCR is a widely accepted and respected method, it relies on an internal calibration curve for quantitative analysis. Therefore, it is imperative to have a reliable reference material.
To assist the AAV field in the qPCR analysis of gDNA, USP and American Type Culture Collection (ATCC) developed reference material for gDNA from HEK293 and Sf9 (Spodoptera frugiperda [army worm]). These cell lines were specifically chosen as they represent the two most common cells used in AAV manufacturing.
Residual Plasmid DNA Reference Material
Removal of DNA impurities, including those from plasmid DNA, is typically performed during downstream production following endonuclease treatment, which is intended to remove accessible DNA fragments. The risk of residual plasmid comes from its ability to get packaged in the AAV capsid and subsequent unintended immunogenic expression. Studies have shown up to 8% of packaged vector genome DNA is composed of plasmid DNA impurities.11 This unwanted DNA, which can cause inflammatory immune responses, is a significant safety risk for patients.12 Residual plasmid is usually assessed via a PCR method that targets known regions of plasmid(s) used in production. Antibiotic resistance gene markers are common target regions for these PCR methods and the primary focus of physical reference material designed by USP.
Fig. 2

USP is developing analytical reference material that can be used for the assessment of residual plasmid DNA. This reference material will be composed of linearized plasmid DNA that contains both the ampicillin resistance and kanamycin resistance (NeoR/KanR) genes. These regions are common target regions for residual plasmid PCR methods. The USP standard will be well characterized via multiple analytical methods, including dPCR. This reference material can be used as either a standard curve calibrant in qPCR assays or as a positive control in either qPCR or dPCR assays.
Genomic Titer Reference Material
Genomic titer is a CQA that is routinely performed during both in-process and release testing. Accurate quantitation of the AAV transgene is vital for proper dosing, as well as for proper characterization of the AAV product. QPCR remains the most common method for genomic titer, but dPCR has emerged as a strong alternative that offers many benefits compared to qPCR, including the ability to perform independent quantitation without a calibration curve.12 Plasmid DNA, which can be quantified by UV-vis, is most often used as a standard for qPCR; however, it is widely reported that using a plasmid standard for qPCR calibration curves does not give accurate results when quantifying the transgene in AAV samples due to the complex secondary structure of the AAV transgene.13,14 USP is developing a genomic titer reference material composed of AAV, not plasmid DNA, because full AAV capsids should be a more accurate standard for the quantification of the AAV transgene.
Fig. 3
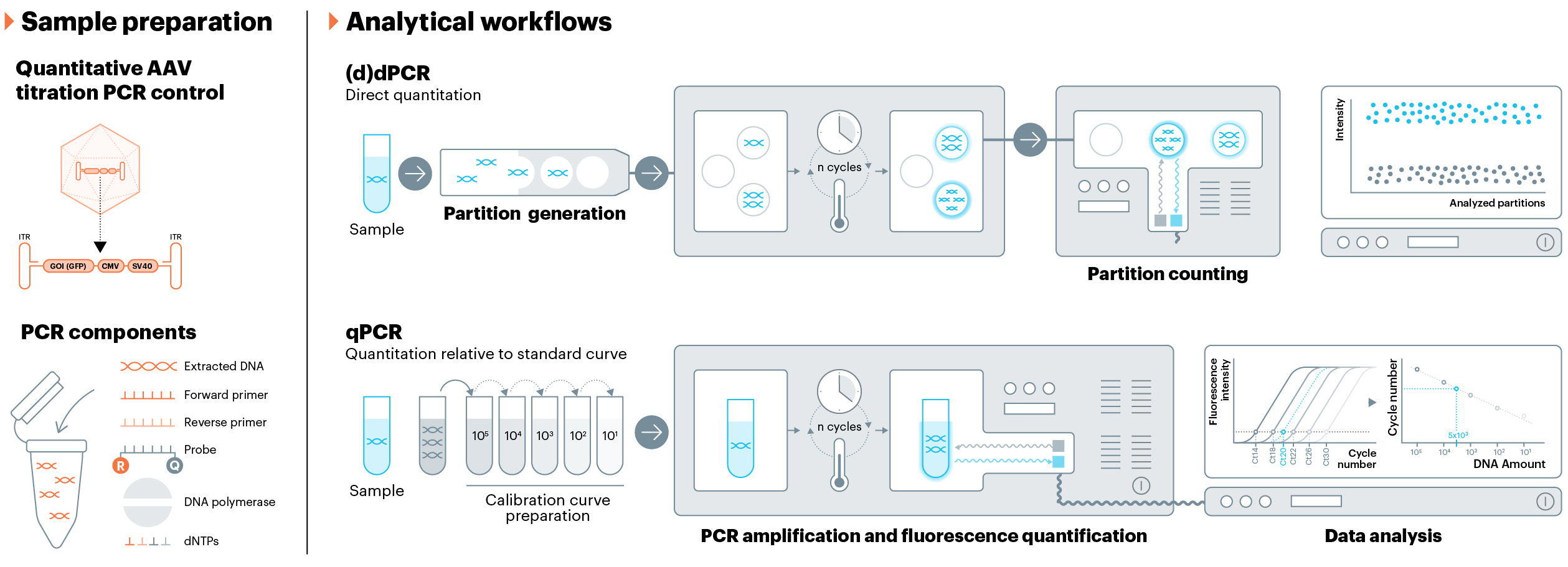
USP reference material will be an AAV (full capsid) with a well-characterized transgene. For the reference material to have universal applications, it must contain conserved regions found in most, if not all, AAV transgenes, such as CMV, SV40, and/or ITR.25 Because ITR is known to be an ill-suited target region, USP will quantify the reference material using the SV40 region and provide a range, which was determined as part of a multi-lab collaborative study.23,24 Similar to the plasmid reference material, this can also be used as either a standard curve calibrant in qPCR assays or a positive control in either qPCR or dPCR assays.
Other Tools For AAV End Users
Along with the physical reference materials being developed, USP is also developing other resources, specifically a USP General Chapter for AAV Best Practices for the Manufacture and Quality Control of Recombinant Adeno-Associated Virus Gene Therapy Products and an interactive online AAV Analytical Guide, that can assist AAV manufacturers. These resources are being developed to better harmonize the AAV field and to provide a centralized AAV hub for both new and experienced scientists in the AAV field.
The general chapter describes best practices for the structure, manufacture, and quality control of recombinant adeno-associated virus (rAAV) products that are intended for in vivo gene delivery applications. The AAV Analytical Guide provides users with a place to browse all the different analytical methods used in AAV characterization and provide useful USP resources, where applicable.
The AAV Expert Panel, which is composed of members from industry, academia, and regulatory, initiated work in June 2022 and has been actively working on drafting a USP General Chapter that provides a general overview of the AAV construct design, along with the control strategy for their manufacture and quality control at small- or large-scale production. A draft of this chapter is expected to be published in USP-PF (Pharmacopeial Forum) in 2025, at which time it will be available for public comments. Following a 90-day public comment period, the expert panel will begin meeting again to review and reconcile the comments. After this task is completed, a final draft will be sent to the expert committee for approval. Pending approval, the final version will be published in USP-NF.
Lastly, USP recently launched an AAV Analytical Guide, which is a webpage designed to provide a comprehensive overview of USP quality and safety resources for AAV gene therapy developers and manufacturers. This web-based interactive tool allows users to explore analytical techniques used across the entire production process, from raw and starting materials to release and stability. Each of the different modules offers users the resources for a variety of quality attributes for AAV analysis. These resources include a list of commonly used methods, as well as USP reference standards and physical standards, where applicable, that are associated with each analytical method.
The USP analytical guide can be found at https://genetherapyanalyticalguide.usp.org/.
Conclusion
As has been highlighted, there are many areas where AAV standards can be used to improve the quality and safety of AAV drug products. USP is working diligently to address these obstacles in a variety of ways. Through the release of physical reference standards or materials and documentary standards, as well as through other learning endeavors, USP will provide a number of help options that can aid AAV manufacturers from production to release testing. Moreover, USP will continue to engage with stakeholders to identify additional bottlenecks and work on developing new standards to address these areas of need.
References:
- Srivastava A, Mallela KMG, Deorkar N, Brophy G. Manufacturing Challenges and Rational Formulation Development for AAV Viral Vectors [published correction appears in J Pharm Sci. 2021 Sep;110(9):3324.
- Jiang Z, Dalby PA. Challenges in scaling up AAV-based gene therapy manufacturing. Trends Biotechnol. 2023;41(10):1268-1281.
- Werle AK, Powers TW, Zobel JF, et al. Comparison of analytical techniques to quantitate the capsid content of adeno-associated viral vectors. Mol Ther Methods Clin Dev. 2021;23:254-262.
- Wagner C, Fuchsberger FF, Innthaler B, Lemmerer M, Birner-Gruenberger R. Quantification of Empty, Partially Filled and Full Adeno-Associated Virus Vectors Using Mass Photometry. Int J Mol Sci. 2023 Jul 3;24(13):11033.
- Richter K, Wurm C, Strasser K, et al. Purity and DNA content of AAV capsids assessed by analytical ultracentrifugation and orthogonal biophysical techniques. Eur J Pharm Biopharm. 2023;189:68-83.
- U.S. Department of Health and Human Services food and Drug Administration Center for Biologics Evaluation and Research, Human Gene Therapy Products Incorporating Human Genome Editing Guidance for Industry. January 2024, https://www.fda.gov/media/156894/download.
- Aucoin MG, Perrier M, Kamen AA. Critical assessment of current adeno-associated viral vector production and quantification methods. Biotechnol Adv. 2008;26(1):73-88.
- US Food & Drug Administration. Guidance for Industry: Characterization and Qualification of Cell Substrates and Other Biological Materials Used in the Production of Viral Vaccines for Infectious Disease Indications. February 2010.
- European Agency for the Evaluation of Medicinal Products. Position Statement on the Use of Tumourigenic Cells of Human Origin for the Production of Biological and Biotechnological Medicinal Products. March 2001.
- Knezevic I, Stacey G, Petricciani J, Sheets R; WHO Study Group on Cell Substrates. Evaluation of cell substrates for the production of biologicals: Revision of WHO recommendations. Report of the WHO Study Group on Cell Substrates for the Production of Biologicals, 22-23 April 2009, Bethesda, USA. Biologicals. 2010;38(1):162-169.
- Wright JF. Product-Related Impurities in Clinical-Grade Recombinant AAV Vectors: Characterization and Risk Assessment. Biomedicines. 2014;2(1):80-97.
- Bucher K, Rodríguez-Bocanegra E, Wissinger B, Strasser T, Clark SJ, Birkenfeld AL, Siegel-Axel D, Fischer MD. Extra-viral DNA in adeno-associated viral vector preparations induces TLR9-dependent innate immune responses in human plasmacytoid dendritic cells. Sci Rep. 2023 Feb 2;13(1):1890.
- Wang Y, Menon N, Shen S, Feschenko M, Bergelson S. A qPCR Method for AAV Genome Titer with ddPCR-Level of Accuracy and Precision. Mol Ther Methods Clin Dev. 2020;19:341-346.
- Prantner A, Maar D. Genome concentration, characterization, and integrity analysis of recombinant adeno-associated viral vectors using droplet digital PCR. PLoS One. 2023;18(1):e0280242.
- Werling NJ, Satkunanathan S, Thorpe R, Zhao Y. Systematic Comparison and Validation of Quantitative Real-Time PCR Methods for the Quantitation of Adeno-Associated Viral Products. Hum Gene Ther Methods. 2015 Jun;26(3):82-92.
About The Author:
 Anthony Blaszczyk is in the Pipeline Development group within USP’s Global Biologics department. At USP, he works with scientific experts and stakeholders to develop new standards to support biopharmaceutical quality assessment and development. Prior to USP, Anthony worked at Catalent Cell and Gene Therapy, where he managed an analytical development team responsible for the development, qualification, and transfer of methods. He obtained his Ph.D. in biochemistry from Penn State University in 2018.
Anthony Blaszczyk is in the Pipeline Development group within USP’s Global Biologics department. At USP, he works with scientific experts and stakeholders to develop new standards to support biopharmaceutical quality assessment and development. Prior to USP, Anthony worked at Catalent Cell and Gene Therapy, where he managed an analytical development team responsible for the development, qualification, and transfer of methods. He obtained his Ph.D. in biochemistry from Penn State University in 2018.