
MHRA Issues New Regulation On Modular And Point Of Care Manufacture Of ATMPs
By Peter H. Calcott, Ph.D., FRSC, president and CEO, Calcott Consulting LLC
Most cell and gene therapy products, including CAR-T therapies, share a commonality whereby the manufacture of the product for the patient is complex and very personalized. Unlike classical biologics where a lot of material can treat hundreds if not thousands of patients, these new products are designed for individual patients or much smaller numbers. What this means is that the efficiency of scale-up cannot be gained. Earlier last year, I wrote an article pointing out this and other dilemmas and posing creative ways to combat it.1
While these medicines do have a component of the manufacturing process which is not personalized (for instance the gene in the vector for gene therapy), the harvesting of patient cells, their treatment or transfection and growth to administrable quantities has to be performed at the patient level. That means that regulators have considered the product to not just be the vector, but also the treatment of patient cells. The product release is moved from the factory to the bedside. Thus, each person’s treatment is considered a batch to be released. Regulators have considered these activities at the bedside to be part of the manufacturing process (Figure 1).
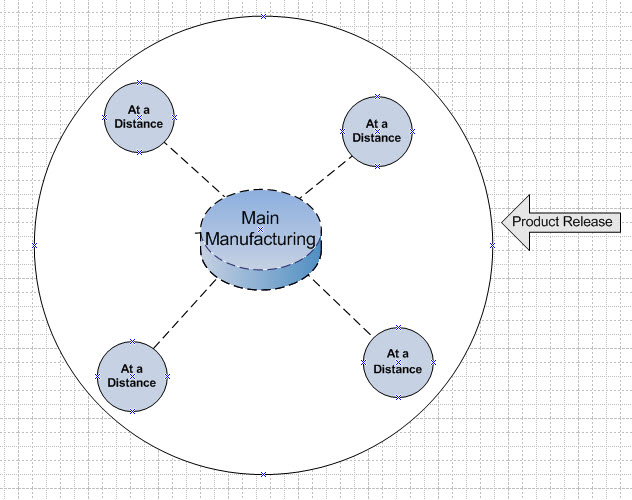
Figure 1: Conventional Model – product release is at the bedside. “POC” = Point of Care.
Conventional biopharmaceuticals and certain injectable non-biologicals also have bedside preparations. Often, the freeze-dried product is reconstituted with water for injection or for perfusion. But this is so commonplace that regulators do not consider this part of the manufacturing process. So, drug manufacturers provide instructions for physicians or patients on how to prepare these drugs bedside or simply at the point of use. In my article, I indicated that it would be wonderful if we could do the same thing for these new treatments. That is to simplify or standardize the process for cell harvest, treatment, and proliferation, so that It could be performed by a process as accepted as bedside preparation of classical drugs.
It appears that the MHRA (Medicines and Healthcare products Regulatory Agency) of the U.K. has done just that. Initially in a release in October 2024, the agency outlined the proposed strategy.2 In January of 2025, it issued an amendment, changing the existing drug regulations3 to allow for a creative way to simplify operations. It will become effective six months after issuance, becoming effective on July 23, 2025. The new regulation, U.K. Statutory Instruments 2025 No. 87, – MEDICINES – The Human Medicines (Amendment) (Modular Manufacture and Point of Care) Regulations 2025, describes a regulatory pathway that will bring relief to the industry.4 The MHRA has coined the term “decentralized manufacturing” to describe the activities that are part of the manufacturing process that are performed outside the conventional factory. This legislature was penned in consultation with 16 different regulatory bodies via the International Coalition of Medicines Regulatory Authorities (ICMRA).
In its model, the MHRA describes two different situations: modular manufacturing (MM) and point of care (POC) manufacturing. MM is where manufacturing activity is performed away from the classic manufacturing site, perhaps at a clinic or hospital laboratory. POC is where the activity is performed very close to the patient or at the bedside, again perhaps in a clinic or hospital setting. Depending on the product and how it is used, one of the models will be the appropriate choice. Part 2 of the regulations describes the commercial product process. In the discussion that follows I will be describing them interchangeably as they are very similar in how the regulatory body is planning on handling them.
In both models, the MHRA is using the Drug Master File (DMF) process. Ordinarily in Europe, DMFs are used to register a bulk substance with the agency by its manufacturer to be referenced and used by the final drug product manufacturer who uses that bulk drug substance (it is very similar to the DMF used by the FDA in the U.S.). In this model, the agency has created two new licenses for medicinal products: “manufacturer’s license (MM)” and “manufacturer’s license (POC)”. The holders of these manufacturing licenses are described as the “control site,” meaning that it has responsibility to supervise and control the sites executing the final manufacture or assembly of the product for the patient. These secondary sites that perform the final stages of manufacture or assembly of the product will be licensed through the Master File (MF) process. The license holder of the manufacturer’s license (MM) creates the MF, which is followed by the secondary site. Thus, the product release is performed at the centralized manufacturing site, the holder of the manufacturer’s license (MM). The agency opines that for reasons relating to deployment, the licensing authority determines it necessary or expedient to be manufactured or assembled in a modular unit, perhaps driven by a short shelf life or the personalized nature of the treatment. This modular manufacturing unit is potentially relocatable.
The same principles hold for point of care products. In this case, the manufacturer’s license (POC) would be responsible for creating the MF, and for supervising and controling the secondary sites. The POC model applies for reasons relating to method of manufacture, shelf life, constituents, or method or route of administration, and can only be manufactured at or near the place where the product is to be used or administered. Figures 2 and 3 illustrate these scenarios. Product release occurs at the main manufacturing site only.
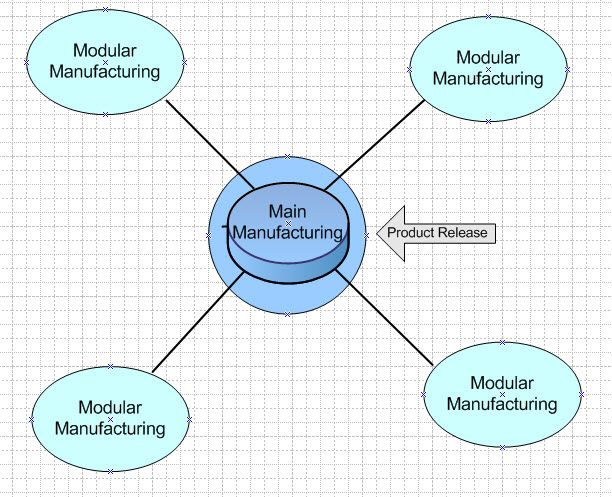
Figure 2: Modular Manufacturing Model – product release occurs at the main manufacturing site.
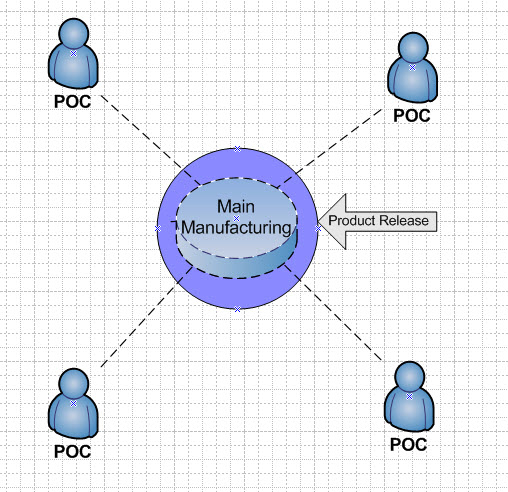
Figure 3: Point of Care Model – product is released at the main manufacturing site.
Approval of these products requires a manufacturer’s license (MM or POC) and an appropriate MF with full instructions on manufacture and/or assembly of the product at a distance. The dossier to be submitted must also describe the satellite sites to be used. This becomes the submission that is evaluated and approved. After approval, the license owner is responsible for assuring the satellite sites are operating according to the MF. If new sites are to be used, they become the subject of a further submission to the agency. The relationship between license holder and satellite sites is very similar to that of license holder and CMO where manufacturing is outsourced. The product made by the satellite is still the responsibility of the license holder.
The onus of responsibility for the product rests squarely on the shoulders of the manufacturing license holder (MM or POC). That means responsibility for generation of an MF (to be submitted with the application) as well as supervision and control of the satellite locations. As experience increases and changes are needed to the MF, the manufacturer’s license holder is responsible for updating the MF as needed. This updating will be as simple as that for a DMF and will not require the manufacturing license to be resubmitted or a variation written for it.
As expected, each product under question will have a separate license and MF, which will be product-specific. Sites must hold a specific license (mentioned in the manufacturer’s license) and follow the specific MF. In simple terms, there is no mix and match possible. The holder of a manufacturer’s license (MM) must ensure that the medicinal products specified in the license are not handled, controlled, stored, or distributed on any premises other than the MM control site and the modular units specified in the MM master file. Just like with a conventional license, changes to the process or facilities are governed by standard regulations. The license holder must assure that manufacturing is performed according to the registered process in the dossier and in the MF. The regulation goes into detailed expectations with respect to the submission dossier content.
As with conventional pharmaceuticals, a pharmacovigilance program must be in place. The manufacturer’s license holder (MM or POC) is responsible for the collecting and processing of data and generation of reports. The standard expectations for conventional products are required for ATMPs. Appropriate vigilance should be paid to assure efficacy and product properties are consistent: these should be reported as appropriate. Pharmaceutical labeling is expected and similar to conventional drugs. However, if the POC medicine is used immediately after assembly or manufacture, it is exempt from the labeling requirements.
Annual reporting for these two types of products is as expected, with regular updates to the license and MF expected also. There must be in place a description of the processes by which the proposed holder of the authorization will initiate, suspend, and cease manufacturing or assembly of the product at an MM or POC location.
In Part 3 of the regulation, there is a detailed description of how these changes impact clinical products (investigational medicinal products [IMP]). In this case, the licenses include “(IMP)” in their designation, e.g., manufacturer’s license (IMP) (MM). The same principles apply to clinical products as described for commercial products. While most elements are the same, there are some minor subtle differences. I advise you to review the regulations for these small differences.
As pointed out by the MHRA, it is the first regulatory body to tackle the challenge of this innovative new area of medicine. Its implementation will be monitored over the next few years not only by the MHRA but I am sure by other regulatory bodies, including the 16 that were part of the process at ICMRA. This creative way of managing these types of products will help to reduce the costs of these products in the long term, moving product release from the bedside back to the factory.
New as of March 19, 2025: The MHRA has posted a new webpage to help industry understand the new regulations.
References
- Strategies to Tackle CAR-T Product Challenges. 2024 https://www.cellandgene.com/doc/strategies-to-tackle-car-t-product-challenges-0001
- Statutory Instrument laid in Parliament provides first regulatory framework of its kind that will transform the manufacture of innovative medicines at the point of patient care https://www.gov.uk/government/news/statutory-instrument-laid-in-parliament-provides-first-regulatory-framework-of-its-kind-that-will-transform-the-manufacture-of-innovative-medicines-at
- MEDICINES – The Human Medicines Regulation 2012 No.1916 https://www.legislation.gov.uk/uksi/2012/1916/contents/made
- MEDICINES - The Human Medicines (Amendment) (Modular Manufacture and Point of Care) Regulations 2025 No.87. https://www.legislation.gov.uk/uksi/2025/87/contents/made
About The Author:
 Peter H. Calcott, D.Phil., is president and CEO of Calcott Consulting LLC, which delivers solutions to pharmaceutical and biotechnology companies in the areas of corporate strategy, supply chain, quality, clinical development, regulatory affairs, corporate compliance, and enterprise e-solutions. He has also served as an expert witness. He also teaches at the University of California, Berkeley in the biotechnology and pharmaceutics postgraduate programs. Previously, he was executive VP at PDL BioPharma, chief quality officer at Chiron and Immunex Corporations, and director of quality assurance for SmithKline Beecham and for Bayer. He has also held positions in R&D, regulatory affairs, process development, and manufacturing at other major pharmaceutical companies. He has successfully licensed products in the biologics, drugs, and device sectors on all six continents. Calcott holds a doctorate in microbial physiology and biochemistry from the University of Sussex in England. He has been a consultant for more than 20 years to government, industry, and academia.
Peter H. Calcott, D.Phil., is president and CEO of Calcott Consulting LLC, which delivers solutions to pharmaceutical and biotechnology companies in the areas of corporate strategy, supply chain, quality, clinical development, regulatory affairs, corporate compliance, and enterprise e-solutions. He has also served as an expert witness. He also teaches at the University of California, Berkeley in the biotechnology and pharmaceutics postgraduate programs. Previously, he was executive VP at PDL BioPharma, chief quality officer at Chiron and Immunex Corporations, and director of quality assurance for SmithKline Beecham and for Bayer. He has also held positions in R&D, regulatory affairs, process development, and manufacturing at other major pharmaceutical companies. He has successfully licensed products in the biologics, drugs, and device sectors on all six continents. Calcott holds a doctorate in microbial physiology and biochemistry from the University of Sussex in England. He has been a consultant for more than 20 years to government, industry, and academia.