
Managing Change In Vaccine Production With All Stakeholders
By Eric Doerr, Manufacturing Sciences and Analytical Technology, Sanofi, Toronto

It is without question that the road toward full implementation, submission, and acceptance of a new product, technology, unit operation, and/or other significant process change within the scope of biopharmaceutical manufacturing is very rarely a straightforward one.
The change itself, from the current to the proposed state, requires clear, concise communication in order to be made effective. It requires definition for causal/effect relations, rationale for implementing the change and proof of impact/risk mitigation, and effective monitoring of the post-implementation state to assure product quality and process performance remain within the validated state.
However, managing the expectations and opinions of different stakeholders, coordinating their efforts, and sometimes mitigating their disagreements, can prove even more daunting than the improvement of the product or process itself.
This article, the first in a two-part series, aims to provide you — whether you are a future innovator, a change manager, a process scientist, or any other member of the bioprocessing industry — with a toolkit to manage the many stakeholders along the road to full implementation of the process or product you wish to advance. Using five real-world case studies from the field of vaccines manufacturing, with considerations based on industry guidelines, technical expertise, and advancements in digitalization, this overview of process change implementation will highlight which skills should be part of your own validation toolbox and will hopefully help accelerate and simplify the road to process change implementation in a straightforward manner.
Guidelines For Continuous Improvement
The International Council for Harmonization of Technical Requirements of Pharmaceuticals for Human Use (ICH) defines continuous process verification, or CPV, as “an approach to process validation that includes the continuous monitoring and evaluation of manufacturing process performance.” Across a product’s or process’ life cycle undergoing improvement or validation, there are five major stages as identified in the figure below that can be further sequestered into three considerations when prepping both your process and the expectations of the major process stakeholders.
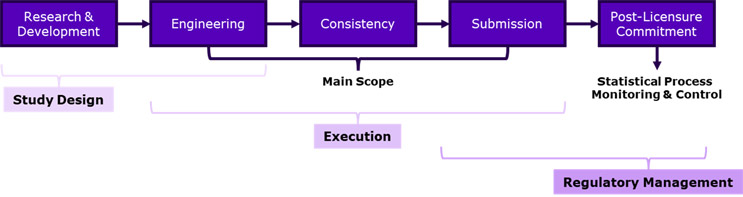
At the start, during R&D and early engineering phases, the definition of the study design is critical for establishing the exploratory design space and identifying where opportunities/risks exist. Establishing the study’s positive and negative controls is essential to provide evidence of the null hypothesis the process improvement aims to achieve and also helps establish the baseline to assess progress from the current to the proposed state. The integration of a design of experiments (DOE) approach with batch-to-batch design strengthens the knowledge base going into and going through process improvement as well. Clearly defining the scientific or technical boundary conditions, as well as establishing early the normal operating ranges and/or proven acceptable ranges (NORs and/or PARs), builds the technical story to keep understanding on par between stakeholders.
Of course, the main scope of these articles — and with the achievement of the end-to-end process improvement definition — is between the engineering, consistency, and submission phases as defined by execution. The previous phase was largely focused on defining the technical and scientific conditions of the process; the next phase’s criticality is dependent on the boundaries set by the team, the stakeholders, and the authorities for approval acceptance.
Clear-cut and consistent role/responsibility definition enlists the person to the task and eliminates ambiguity from the many tasks and sub-tasks required to carry a process improvement from start to end. Prior to the actual execution start, it is critical to centralize the data with effective archival/databasing techniques in one chief location. Limiting step-to-step changes or transformation while the data moves from raw data file to analysis to reporting reduces the likelihood of transcription errors and/or misinterpretation along the validation life cycle. Further to this, the ability to report interim results live is streamlined, thereby presenting up-to-date data and information to team members in the hope of further optimization, troubleshooting diagnostics, and/or delivery of major project milestones.
These interim results and databasing techniques also assist with the accelerated development of final submission reporting, significantly shortening review/approval cycles (an especially helpful benefit with the number of reviewers these documents may have!).
The final aspects of process improvement to consider at the submission and post-licensure commitment phases largely involve appropriate and succinct regulatory management. End-stage validation runs should involve associated regulatory affairs and/or legal departments to ensure the language in these reports matches the medical-legal requirements typical of a submission dossier, specification, and/or monograph.
This preparatory review also ensures that the commitments made for licensure continue to be supported by the results of the process performance qualification report. Any discrepancies to be resolved internally can be handled in a common language, and for those discrepancies that may pose a risk for the completion of the project, open and concise exchange with the applicable health authorities should be made to determine what must be delivered for submission approval and what can be covered as a post-licensure commitment instead.

When Small Changes Could Bring Big Benefits … And Huge Compliance Hurdles
A centrifugal process step included as part of the downstream purification of a product of interest was identified as the major rate-limiting step in terms of product recovery. Small-scale studies using representative process intermediates determined that changes in centrifuge design and operational parameters could result in a 30% to 50% increase in recovery at the drug substance stage. However, said improvements had major regulatory and compliance hurdles to meet, including the requirement to perform full process validation on a long and highly complex purification stream, establishing cross-comparability between two manufacturing facilities producing this product from the current to the proposed state, and addressing the learning curve for the newly acquired equipment and consumables with respect to procedures, handling, and robust operational understanding.
The first major tool to ensure that the process validation was well understood and that the current execution status could be clearly/accurately communicated was the development and routine use of a live execution data dashboard. The ability to meet with stakeholders on a routine basis to highlight the process’ expected behavior, or even more importantly, to meet ad hoc when an unexpected event or excursion occurred, allowed for rapid root cause identification.
To assist the team with ensuring process and product knowledge was retained/improved upon across the validation process, the testing strategy and verification of the explored design space at both the centrifuge operation and subsequent purification stages was the second tool considered, as up-front, defined exploration and definition give stakeholders the confidence that any change is behaving as expected without deleterious concerns.
Finally, the execution team built redundancy into the protocol for successful execution and worked with the manufacturing area’s stakeholders to secure seamless action if a process failure and/or an inability to secure materials were to occur during execution.
Using the above tools, the process was successfully validated, implemented, and accepted by all health authorities, with a comparable and improved drug substance used throughout all drug products where required. The new process continues to surpass the yield targets established by working within the validated design space for optimal recovery.
What Happens When Validation Overlooks Process Failure Modes?
However, once the new centrifugal process was validated and established within the routine manufacturing environment, there were recurring bottle breakage events during routine manufacturing that were believed to have been mitigated as part of the close-out actions of the validation stage. The challenges to overcome in order to secure the new process in a safe and reproducible manner included the potential product loss – thus jeopardizing the gain in product recovery made possible with the change in the centrifugation process step – the potential for equipment malfunction resulting in subsequent biological contamination within the processing area, and a risk of running routine manufacturing with a process that was unknown in terms of its stability and batch-to-batch predictability.
Investigation began with performing core team walkthroughs of the manufacturing space during centrifugation by operational, technical, and quality stakeholders for a multidisciplinary review of current practices and procedures. This not only utilized the many talents and perspectives of the core team but also facilitated unanimous alignment on how the process was performed without lapses in memory and/or opinion. These observations and results were then tabulated and presented under the guidance of the next tool – the performance of a failure modes and effects analysis (FMEA) workshop.
The FMEA exercise takes all known inputs/outputs of the process to see what could be contributing to a manufacturing failure or defect, such as the broken bottles, and identifies solutions typically outside of the day-to-day activities that operators face. Following the conclusion of the FMEA, consultation with a different set of external subject matter experts (SMEs), an independent third party in fact, provided clarity to identify the root causes plaguing the many successes the new process created. It is critical to ensure the right SMEs are identified and participate with manufacturing investigations as early as possible to accelerate root cause identification and de-risk daily operations.
Ultimately, the definitive root causes of the recurrent bottle breakage events were identified, and with effective corrective and preventive actions/effectiveness reviews (CAPA/ER) management, risk/impact assessment securing product quality metrics, and long-term process monitoring under a quality-approved protocol, the structural integrity of the centrifuge bottles has remained in place with no recurrence since.
 About The Author:
About The Author:
Eric Doerr is a senior project lead for upstream and downstream processing areas at Sanofi where his work supports and improves global vaccine supply chains. He earned his bachelor’s and master’s degrees in biochemical engineering science from the University of Western Ontario.