
7 Key Areas Of Risk In Single Use Bioprocessing — Is Your Process Protected?
By Andrew Kelly and Graeme Proctor
Your biologic drug saves lives, but only if it is available when and where it is needed. So, how can you ensure it is? A strong risk mitigation strategy is necessary. There are seven areas of risk associated with single-use technology. We will summarize each area, and suggest strategies for ensuring patients receive your contaminant-free drug, on time.
INTRODUCTION
Single-use technology (SUT) offers many well-documented benefits. But, unlike its long-lasting stainless-steel predecessors, SUT is disposable. Therefore, reliable replenishment is needed. Without the disposable equipment you cannot make your product.
On-time delivery of the supply is not the only risk. You also must trust its reliability and quality. SUT users need a comprehensive risk mitigation strategy to address the selection, qualification, and use of consumable processing equipment and materials used for biopharmaceutical manufacturing. Additionally, the strategy must address risks related to hardware, operational procedures, and the supply chain.
This guide addresses risks associated with the specific application areas, technical challenges, operations, and supply chain, as well as mitigation strategies for both the drug manufacturer and the equipment vendor. By protecting your process from potential risks, you protect patients by ensuring a steady supply of lifesaving drugs.
1. UPSTREAM TECHNICAL RISKS
Although in upstream manufacturing, the target molecule may still be impure and unrefined. Problems at this stage are still liable to affect downstream product quality, create delays in production, and ultimately impact delivery of medicine to patients. Sources of upstream technical risk include:
Raw Materials
Variation in the quality of incoming raw materials, such as media components and other bioreactor additions, may influence the process yield. There is also potential for introduction of contamination through adventitious organisms or viral particles present in the source material that can lead to declines in cell productivity.
To reduce risk of contamination at this stage, it helps to take a holistic view with regard to incoming materials and identify areas where changes could yield improvements. For instance, is it possible to mitigate contamination risk by switching to the use of a chemically defined growth media source rather than a serum-based media option?
Hardware Cleaning & Maintenance
Scheduled service, maintenance, and calibration of hardware systems used to monitor and control processes is critical to reduce process variation. In addition, the use of validated cleaning and sterilization protocols is critical to avoiding contamination of product contact components where single-use flow paths are not in use.
Preventing Bioreactor/Product Contamination
Incoming bioreactor feeds, whether liquid or gas, should be either bulk sterilized or sterile filtered prior to introduction to the system in order to control contamination risk. Where appropriate, mycoplasma retentive filtration steps may also be employed on incoming media feeds at this stage.
Intermediate Storage & Transportation
Any intermediate product storage stages should also be appropriately validated, together with the equipment and process conditions utilized, in order to mitigate the risk of contamination, consumable failure, or potential influence upon other aspects of product quality.
2. DOWNSTREAM TECHNICAL RISKS
As the product moves through the various stages of downstream processing, its overall value and the concentration of the drug molecule solution both increase significantly. This means any loss during these stages will be extremely costly both economically and for continuity of supply. Areas of downstream technical risks include:
Process & Process Stream Variation
Inherent variation in any process system operation may result in variable product quality. It may be possible to reduce this by implementing appropriate system automation around processes such as TFF, chromatography, and virus filtration.
Additionally, variation of the process stream itself can create problems around downstream filtration and chromatography steps if the levels of components such as cell debris and nucleic acid are higher than typically expected. Elevated levels of antifoam compounds, used to control foaming during upstream fermentation, will also significantly impact upon these processes if not carefully limited.
Leachables
Leachables from polymeric product contact surfaces also become more significant as the drug moves through downstream processing steps. Recent industry focus on standardized extractables protocol is likely to offer a greater volume of highly detailed vendor data, but this data will be complex and must still be thoroughly assessed using the appropriate toxicological methods.
Contamination
Avoiding contamination becomes even more critical as the drug molecule gets nearer to the intended patient. Selection of appropriate filtration technology is vital to ensure process sterility and remove potential microbial and viral contamination from the drug itself, but additional process steps must be designed so as not to create alternative risks. For example, in vaccine production, use of closed-loop, pre-sterilized tangential flow steps during ultra-filtration processes may significantly reduce the risk of exposing the product to external contamination.
Storage & Transport
Storage and transport of the drug should not be overlooked. Whether it is bulk-filled product being shipped for further processing or the final packaged product, the container design and functionality can protect against damage, subsequent contamination, and lost product. An extremely well-designed and controlled process will of course eliminate a large proportion of potential risk. However, this could well be rendered irrelevant if the strategy does not extend to protection of the drug once it leaves the manufacturing facility.
Product Stability
Similarly, product stability programs are critical to ensure the drug remains safe and viable throughout its usable lifetime, when transported and stored under appropriate conditions.
3. NON-TECHNICAL RISK SUMMARY
Although risks directly affecting the production processes used in drug manufacture are extremely important, not all problems arise from technical failure or product contamination events. Consider the impact of a disruption to the supply chain or the manufacturing and related sites.
A manufacturing site can be affected in many ways. Natural disasters (e.g., earthquakes, volcanic eruptions, tropical storms) can significantly disrupt transport or sites but are limited to specific geographical locations. However, it is possible for weather events such as heavy snow or torrential rain to significantly disrupt transport and utilities, which may play a key role in maintaining a manufacturing facility.
Disruption of the supply chain can extend beyond the arrival of raw materials and consumable equipment on site. Employees may be unable to travel to the manufacturing site, which can have an equally significant impact upon production.
Ultimately, it is impossible to remove the potential for some of these events to occur. However, plans can be put in place to limit the impact on production should such a situation arise. In these scenarios, it is also important to consider risks throughout the critical supply chain in addition to the manufacturing site.
The Icelandic volcanic eruption in 2010 is a good example of an event that created significant transport interruption across a wide geographical area. In an instance like this where air transport is affected, those with alternative land and sea transport contingency plans would experience far less disruption.
Planning for an unexpected disruption may well be the difference between patients having access to lifesaving drugs or not.
4. AVOIDING CONSUMABLE FAILURE
Although consumable components are typically drawn from a standard pool of options, the wide range of materials and designs available means selection must be carefully considered. It is often advantageous to seek the assistance of the relevant vendor.
Typical operating parameters should be considered during validation, but potential deviations in these areas must also be understood. For example:
- Is the optimal tubing or element material being used for welding, pumping, or pressure-resistance requirements?
- Has filter retention performance been appropriately validated at the limits of process conditions?
- Have system test requirements been considered, and will they require modification to the system assembly if flushing or pressurization is required?
- Are consumable stocks maintained to ensure they are always used within their validated shelf life?
QUALIFICATION OF CONSUMABLES
Ideally, the qualification of consumable components should begin as soon as possible during a drug’s lifetime. Scaling-up a process is likely to be significantly easier if appropriate consideration to process operation and suitable component selection is applied at an early stage.
It is usually beneficial to seek equipment vendors’ opinions on selection. They can offer expert advice on the performance of their products under the specific conditions in a given process.
Physical and chemical operating parameters must be reviewed. Is the component designed to withstand exposure to the pressures and temperatures expected in the process or during system sterilization? Are there any chemical compatibility concerns around material selection? Additionally, exposure time must be factored into these decisions.
Filtration components should be selected to offer the required level of retention, and any integrity testing requirements should also be factored in. But, it is also important to understand what impact any variation in the physical characteristics of the process fluid may have. If cell or protein aggregation were to occur, what impact would this have on a filtration step, and would it prevent a full batch from being processed?
Leachables analysis should also be performed. With the introduction of new and comprehensive standard extractables testing protocols, the data available from vendors should typically provide model solvent analysis to fit most common application requirements. But, suitable risk assessment processes must still be conducted to analyze and understand the data in relation to the drug and patient.
Vendors can also often provide assistance with development and performance of a suitable validation strategy, facilitating the outcome of providing a highly robust process with minimal risk of consumable failure.
NORMAL FLOW FILTRATION
Normal flow filtration plays a central role in maintaining the quality of drug products. The selection of an appropriate filter for the product stream at any given point will primarily be driven by the level of retention needed.
In some circumstances, a highly retentive filter may be required to remove diminutive organisms, such as mycoplasma, from process fluids. However, implementing an extremely retentive filtration step where it is not required will only create additional burden on the process. Therefore, it is important to understand precisely what is appropriate at each stage.
Sterilizing-grade filters are frequently employed, and correctly so in many aspects of biopharmaceutical production. But to employ a sterilizing-grade filter where only bioburden control is needed requires an oversized filter stage, may restrict the process, and will create the need for additional filter-integrity testing steps.
In many cases downstream processing stages are not truly sterile. Chromatography is a prime example. Therefore, in this situation a bioburden control filter will provide sufficient process protection without risking unnecessary additional process operations.
Integrity testing protocols should be designed to provide assurance of filter integrity but must also be balanced against introducing further process risk. Additional flushing and testing operations may confirm the filter is still fit for purpose but can also risk introducing contamination to the system. From a product safety perspective, the data provided may be redundant when further post-use integrity testing is performed anyway.
Vendor assistance during filter sizing exercises is also recommended. The filter supplier may offer additional experience in relation to a specific filter and application pairing. Vendor expertise can go beyond data from a single bench-top capacity study and may help to account for the effects of long-term process variation upon filter performance.
Operating and testing procedures should also be designed to deal with troubleshooting filtration operations. If a filter blocks prematurely due to process variation or if an integrity test fail result is returned, are these eventualities written into the operator protocols? Has the vendor provided input regarding best practices in each case?
TANGENTIAL FLOW FILTRATION
When utilizing tangential flow filtration, the membrane cut-off used to achieve the appropriate degree of retention or transmission is clearly important.
However, it is always good practice to ensure the process has been optimized to provide repeatable performance under defined operating conditions. For example, faster processing or better membrane recovery can be achieved through optimization studies.
Module format should be considered. Cassettes and hollow fibers are commonly used but may provide advantages in certain situations arising from process needs.
In some applications, aseptic closed-loop processing may be highly beneficial. For example, some vaccines are too large to be sterile-filtered, and therefore processing in a pre-sterilized single-use system is ideal. This means gamma-stable cross-flow elements with very low extractable content are extremely beneficial.
Sterilization methods should also be considered. Is gamma irradiation used? If so, a product that can withstand gamma irradiation is required. Or, if autoclave sterilization or caustic sanitization will be implemented, autoclavable or reusable elements would be appropriate.
In any application, process systems should also be designed to maximize product yield through the elimination of unnecessary tubing or pipework volume and minimize any potential for product to be lost within the process.
SINGLE-USE ASSEMBLIES
When implementing single-use flow paths and processing steps, it is vital that the system design and componentry selection match the specific application requirements. Assemblies must be designed to provide robust, repeatable performance from initial delivery, handling operations, installation, processing, and beyond into drug storage, cold chain, and transport. All aspects of use should be considered. In a highly controlled manufacturing environment, a failure may be unlikely. But shipping of drug products is likely to expose containers and closures to increased risk of damage, so they must be designed and transported accordingly.
The potential complexity of single-use assemblies and the number of component types and polymers they contain means extractable and leachable studies are a critical aspect of selection and use. Vendors should be able to provide analysis to support validation risk assessments performed prior to the start of drug manufacture. In fact, it is beneficial if the vendor can adopt an advisory role during assembly design. In addition to design experience, the vendor can offer extensive qualification data on components from within their catalog which can then be incorporated into a robust process validation strategy.
5. AVOIDING OPERATIONAL ERROR
Operational error can result from one of three main areas: consumables and assembly manufacturing, human or process variation, and equipment breakdown and failure.
Consumables & Assembly Manufacturing
Error can creep into the process at this stage based upon how the consumables or assemblies are manufactured. One way to minimize this is to standardize and ensure the SUT is as robust as possible. Single-use flow paths can be assembled in-house but may not be identical from batch to batch due to not making the assemblies day in and day out. This is a possible source of variability or error.
Choosing The Right Retainer Type
Manufacturing techniques and the use of the correct consumables can build robustness into your process. Vendors can help both to standardize solutions and ensure quality and robustness are forefront. The five examples below in Table 1 are retainer options used in single-use assembly manufacturing. The various retainer options described are listed in order of increasing costs.
|
Retainer method |
Description |
|
No retainer
|
No external retainer is the riskiest approach. While it is the cheapest option with no additional cost of retainers involved, it relies on the physical properties of the union between the hose barb and the tubing alone and provides the weakest of all connections listed here. |
|
Tie wrap
|
Available in many sizes and offer many benefits such as being low cost and flexible enough to work with all material types. But they can create a non-uniform compression between the tubing and the hose barb, and the application is operator-dependent and manual. |
|
Oetiker clamp
|
Available in many sizes and offer many benefits such as being low cost and flexible enough to work with all material types. They offer 360-degree compression and an automated tensioning system which can be applied to a known force. |
|
BarbLock
|
Offer 360-degree compression and a repeatable automated connection force while being compatible with all material types. But, they are more limited in sizes, more costly, and sometimes difficult to attach. |
|
Overmolding
|
Overmolding can only join like-for-like materials but gives the advantages of a uniform internal flow path and a single material of construction as it removes the need for a hose barb. It is ideal for applications which require long-term frozen storage because of its single material construction (i.e., the expansion and contraction rates of all components are equal). |
Table 1: Retainer Options Used In Single-Use Assembly Manufacturing
Importance Of Correct Retainer Positioning
While the choice of retainers may be application-specific, some areas of the manufacturing process can make or break the robustness of the assembly.

Figure 1: Importance Of Correct Retainer Positioning For Single Use Assemblies
The images in Figure 1 demonstrate the importance of the correct positioning of the retainer using tie wraps and Oetiker clamps. The dotted yellow lines show the position of the actual hose barb within the tubing.
Both of the tie wraps have been attached away from the hose barb, and the image on the right shows that under liquid pressure, these will move toward the hose barb, and, whilst doing so, the tube will also shift in its position — giving the potential for leaks.
The bottom images in Figure 1 show some Oetiker clamps. One has been attached away from the hose barb, and the other has been attached butted-up to the hose barb. The image on the right shows that under liquid pressure, the one away from the hose barb will move toward the hose barb, and, whilst doing so, the tube will also shift in its position — giving the potential for leaks. On the left, the retainer butted-up to the hose barb does not show any movement, giving more process security.
These images show that with careful controls and methods, robustness can be built into every assembly .
Give the responsibility to the vendor
To ensure quality is built into every assembly manufactured by Parker, the two most suitable connectors have been standardized, providing the most reproducibility, robustness, and reliability within an assembly.
These are Oetiker clamps and overmolding, or a combination of both, providing flexibility throughout the full tubing size range and the ability to offer solutions across a range of operating and storage temperatures. All of our processes and procedures are validated, with leak testing of assemblies being carried out as part of the manufacturing process.
By doing all of this, we ensure we build quality into our manufacturing processes.
Human Or Process Variation
A second source of operational error comes from human or process variation. One way to minimize this is to automate both the process and the control mechanisms.
When implementing a single-use bioprocessing solution, various levels of automation are available, ranging from completely manual steps, through semi-automated, to completely automated unit operations. The level of automation will depend on the step in question and the cost of implementation and ownership versus the technology transfer and operational improvements achievable.
A fully manual process is very labor intensive and operator intensive. Such a process involves manual data acquisition, manual analysis, and manning of workstations, sometimes requiring many operators to look after a single process step. Timing and endpoints can be subjective, and variation can creep in from many different sources such as human error, transcription, and equipment variation. When process standardization is optimal, manual processing is not ideal.
On the other hand, if we look at automation within the single-use sector, it can offer an immense number of improvements to biopharma processes, including:
- It can free up the operator, allowing for safe and effective multitasking while ensuring the safety of processes and products.
- It can reduce the number of operators required for the process tasks.
- The process can be standardized with many single- controlling parameters such as pressure, and monitoring and providing feedback on parameters such as conductivity and temperature.
- It can eliminate many of the errors associated with human interfacing by utilizing standardized, reliable programming to ensure reliability and robustness.
- It can automate data acquisition and analysis, thereby eliminating transcription errors in reports.
Also, when processes are transferred between facilities, there may be differences in the way processing steps are conducted in order to achieve facility fit. The greater the level of standardization, the faster the implementation and validation will be.
Equipment Breakdown & Failure
A third source of operational error is equipment breakdown and failure. One way to minimize this risk is by developing an appropriate equipment maintenance schedule.
It’s not uncommon to hear critiques of preventive maintenance, such as:
“Preventive maintenance costs money, reduces machine availability, and slows down my production. Why should I do that? If the equipment is not working, it’s not making money!” Or,
“It’s easier, cheaper, and less frequent to just replace a part when it breaks!”
In many industries this may be true. But within the biopharma industry, any unplanned downtime of the equipment can have a tremendous impact on not just profitability, but reputation, speed to market, and, potentially patient care due to product shortages.
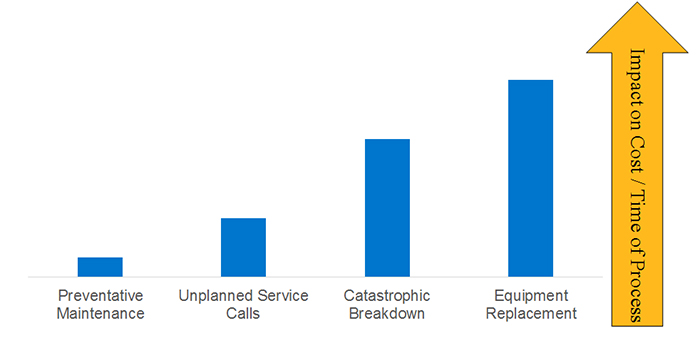
Figure 2: Equipment Maintenance
To avoid such consequences, maintenance planning is essential. In Figure 2 the maintenance type is on the x-axis, and the impact on cost and time due to inability to process is on the y- axis. The least-process-invasive type of maintenance is preventive, where maintenance can be planned around the production schedule. This type of maintenance, while having an ongoing cost, can be the most cost-effective.
The impacts on both processing capabilities and cost sequentially increase through the other maintenance types, with both catastrophic breakdown and equipment replacement having significant effects on the process. This is where lead times for unplanned part failures and total equipment failure can significantly impact the business reputation and product lines. A small investment in maintenance can go a long way to keeping your processes and product lines on track.
6. CONTROLLING CONSUMABLE STOCK
Ensuring your business is able to keep processes running day to day relies on controlling the consumable stock available. In general, monitoring inventory levels is an important part of inventory control. Doing so ensures you never run out of critical consumables, as well as helping to gauge the trends and demands for the items you carry. That, in turn, can help in forecasting, or making an informed prediction about when to place an order.
Forecasting models, such as determining reorder points, can help ensure optimal inventory control. The reorder point is the quantity of inventory that triggers an action to replenish that certain stock. The reorder point answers the question of when to order.
Lead time is a consideration, since instantaneous replenishment of stock levels is not possible. Safety stock offers a cushion. The reorder level is typically calculated as the average daily usage rate multiplied by the lead time in days. This formula assumes a constant rate of inventory usage, which is often not the case. To guard against a stock-out condition, the safety stock level is added to the reorder level. Thus,
Reorder level = average daily usage rate x lead time + safety stock
The key is to have a system in place to always have enough inventories to cover the lead time while waiting for new stock to arrive.
7. DISASTER RECOVERY/BUSINESS CONTINUITY
Thinking about a disaster can put fear into the bravest of people — and this is where disaster recovery or business continuity plans come into play. These plans ensure the business can continue its day-to-day operations without too much pain in the event of a disaster.
A disaster could be natural, environmental, or manmade. Some of the consequences include:
- loss of product or product intermediate
- delay to new products or existing products
- loss of revenue and profit
- negative view of your brand (reputation)
- loss of clients or customers
- product shortages.
Disaster Recovery Planning
Like every insurance plan, benefits can result from generating a disaster recovery plan. Some of these benefits are:
- providing a sense of security
- minimizing risk of delays
- ensuring standby systems are functional
- having pre-approved decision trees available
- reducing potential legal liabilities
- reducing unnecessary stress in the work environment.
How do you develop a disaster recovery plan?
The initial stage is to risk-assess the entire process. This typically involves a cross-functional team within the business to ensure no areas are overlooked. It may also extend to include vendors and contractors based on the criticality of the plan.
Following the process flow, assess each step and rate based upon a meaningful criticality scale for the business involved. Identify the potential hazards (including failure modes) of each step, the consequences of these hazards, and determine the vulnerabilities.
Once the risk assessment is complete, agree upon and document a mitigation plan for each step in the process, answering two key questions:
1. What actions need to be taken should a disaster occur?
2. Who would carry out these actions and take responsibility?
There are no shortcuts to this exercise and it is unlikely you will find a one-size-fits-all plan that meets the needs of your unique business.
Risk Mitigation Strategies
Now that we’ve addressed what the disasters could be and how to plan for recovery from them, what are some strategies to reduce the risk disruption from a disaster or unplanned break in manufacturing?
From a manufacturer’s point of view, risk mitigation strategies include:
- dual sourcing of consumables
- controlling inventory based upon stock levels, usage, and lead times
- having a preventive maintenance plan in place.
From a vendor’s point of view, risk mitigation strategies include:
- having dual sites of manufacture
- having a dual sources of components
- engaging with customers to determine what stock they need to hold
- enabling a fast response time to customer demands
- having their own business continuity plan in place.
While disaster recovery plans can be written in isolation, it is safer and more beneficial for the manufacturer and vendor to work together to minimize any potential risks.
CONCLUSION
To ensure patients receive lifesaving medicines, there are many approaches to process protection. These include eliminating sources of contamination, ensuring continuity of supply and a having a repeatable process. To minimize areas of disruption, business continuity plans are a must for both manufacturer and vendor. Remember, you should always expect the unexpected and prepare for the worst. There is never a good time for things to go wrong when lives are at risk.
About The Authors
Andrew Kelly
Andrew holds a Bachelor’s degree with honors in medical microbiology from Newcastle University, UK.
Having previously held the roles of laboratory scientist and technical support scientist at Parker, Andrew is now product manager for the portfolio of pharmaceutical and life science filtration products, focusing on linking external commercial and marketing demands with the activities of both the engineering and operations business functions.
Graeme Proctor
Graeme is the single-use technology product manager for Parker’s process filtration division based in the North East of the UK. Graeme has worked in the biopharma industry for his entire career. His early work focused on manufacturing antibodies for forensic and analytical testing, followed by some time in academia.
In 1999 Graeme joined a chromatography company. While there, he held R&D, project management and product support roles. He was also responsible for the development and launch of a single-use chromatography platform.
Since joining Parker as single-use technology product manager, Graeme has focused on developing Parker's single-use technology platform for bioprocessing. His current focus is to create robust solutions which will enable customers to improve the quality and accessibility of biopharmaceuticals.
About Parker
As the life science market continues to face challenges with the processing of high value and often fragile products, Parker brings fresh choice and new solutions to meet the filtration and processing needs of the industry.
In addition to a specially designed product range, Parker customers can enjoy a deep commitment and flexibility that is second-to-none. It is these qualities that drive Parker to deliver optimized and tailored solutions to meet each customer’s individual processing needs.
Wherever you are in the world or whatever the requirements of your process, Parker’s dedicated support team are there to help you get the most out of your filtration and fluid handling systems. With multiple manufacturing facilities and a network of customer support centers operating in more than 50 countries worldwide, we can offer a truly global support service.