
Introduction To The ASTM E3219 Standard Guide For Derivation Of Health Based Exposure Limits (HBELs)
By Andrew Walsh, Thomas Altmann, Joel Bercu, Ph.D., Alfredo Canhoto, Ph.D., David G. Dolan Ph.D., Andreas Flueckiger, M.D., Igor Gorsky, Jessica Graham, Ph.D., Ester Lovsin Barle, Ph.D., Ovais Mohammad, Mariann Neverovitch, and Osamu Shirokizawa
Part of the Cleaning Validation For The 21st Century series
This article provides a detailed introduction to the approaches for utilizing preclinical and clinical data to derive Health Based Exposure Limits that toxicologists have been using over the past several decades. It also introduces the approach described in the ASTM E3219 Standard Guide for Derivation of Health Based Exposure Limits (HBELs).1 The procedures outlined in the newly published HBEL standard are expected to greatly aid professionals in the derivation of acceptable levels of carryover and should be used in the risk assessment of cleaning processes using the ASTM E3106 Standard Guide for Science Based and Risk Based Cleaning Process Development and Validation.2
FDA 1996 Proposed Revisions To The GMPs
As discussed in the first two articles of this series,3,4 the Barr Labs court case5 played an important role in the 1996 Revisions to the Good Manufacturing Practices (GMPs),6 which was one of the drivers for the development of the Risk-MaPP7 guidance document. In brief, multiple inspections of Barr Laboratories, Inc. by the FDA resulted in repeated observations. In frustration, Barr Laboratories brought a lawsuit against the FDA. The FDA responded by petitioning the court for an injunction against Barr Laboratories. The outcome of this legal battle was the now famous "Barr Labs decision." In his decision, Judge Alfred M. Wolin found all of the FDA's claims against Barr Laboratories to have merit and agreed that process validation and cleaning validation are required by the GMPs. By extension, the Barr Labs decision applied to the entire industry.
At the same time, Judge Wolin also criticized the GMPs for being vague and not very detailed and sympathized with Barr Laboratories’ complaints about the FDA's apparently capricious and unpredictable enforcement. Partly in response to Judge Wolin's criticisms, in 1996 the FDA proposed changes to the previously established GMPs.8 In these proposed changes, the FDA specified that, in addition to penicillin, certain "classes" of compounds, including "cytotoxic agents or other antibiotics...", would also need to be manufactured in dedicated facilities. In the preamble to the proposed changes, the FDA stated:
"The agency has refrained from establishing a list of drugs or drug products that present such an unacceptable risk, because such a list would quickly become obsolete....... FDA expects manufacturers to identify any drugs that they produce that present the risk of cross-contamination and to implement measures necessary to eliminate that risk. FDA recognizes that, depending on the drug product, a variety of measures may be acceptable to eliminate cross-contamination; there may, however, be situations in which nothing short of dedicated facilities or equipment will be sufficient".
The industry saw many difficulties in complying with the potential dedicated facility requirements, particularly the cost of building facilities for only one product. Compounding this issue was the problem that many companies were already having trouble implementing the criteria for cleaning validation limits published by Fourman and Mullen in 19939 (i.e., 1/1,000 of the therapeutic dose, not more than 10 ppm, and visibly clean), which often resulted in very low limits for low-risk products. If cleaning procedures could not reduce residues to meet these unnecessarily low limits for these low-risk products, then a dedicated facility might need to be considered even for low-risk products.
Risk-MaPP (First Edition)
A team was formed in 2004 to address the challenge of the 1996 GMP Revisions. This team eventually wrote the ISPE Risk-Based Manufacture of Pharmaceutical Products (Risk-MaPP) Guide, which introduced a well-established approach used in determining Occupational Exposure Limits (OELs) for pharmaceutical worker exposure to drugs as the model for assessing the risk of patient exposure from cross contamination in multi-product facilities. The approach described by Risk-MaPP uses the Acceptable Daily Intake (ADI) approach originally developed to estimate safe oral intake amounts of chemicals in food to calculate an Acceptable Daily Exposure (ADE) limit. Risk-MaPP defined the ADE as:
"a dose that is unlikely to cause an adverse effect if an individual is exposed, by any route, at or below this dose every day for a lifetime."
Risk-MaPP (First Edition) was released in 2010 and included the calculation of the ADE limit shown in Equation 1.
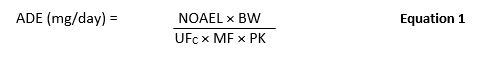
where:
ADE = Acceptable Daily Exposure (mg/day)
NOAEL = No-Observed-Adverse-Effect Level (mg/kg/day)
BW = Body Weight (kg)
UFC = Composite Uncertainty Factor
MF = Modifying Factor
PK = Pharmacokinetic Adjustment(s)
Version 1 of Risk-MaPP was available for a fee and provided approximately six and a half pages of instructions on the derivation of an ADE. This guidance document was helpful in establishing a framework for the derivation of safe levels of contamination. There were, however, still questions from professionals, including questions on the selection and proper usage of adjustment factors (e.g., compound-specific adjustment factors) and modifying factors to account for potential bioaccumulation.
EMA Guideline On Setting Health Based Exposure Limits10
In November of 2014, the EMA released a new guideline
"to recommend an approach to review and evaluate pharmacological and toxicological data of individual active substances and thus enable determination of threshold levels as referred to in the GMP guideline. These levels can be used as a risk identification tool and can also be used to justify carry over limits used in cleaning validation".
Note that the EMA Guideline refers to a "safe threshold value," which is equivalent to the Maximum Safe Carryover (MSC) found in the ASTM E3106. Both terms were coined by one of the authors (Walsh). Many professionals have adopted the MSC as it is a more informative term and an obvious replacement for the Maximum Allowable Carryover (MAC) term.
The EMA's 2014 Guideline included the calculation of the Health Based Exposure Limit (aka PDE) as depicted in Equation 2.
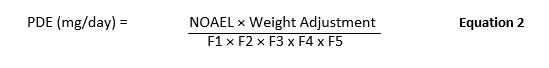
Where:
F1: Factor to account for extrapolation between species
F2: Factor to account for variability between individuals
F3: Factor to account for repeat-dose toxicity studies of short duration.
F4: Factor that may be applied in cases of severe toxicity, e.g., non-genotoxic carcinogenicity, neurotoxicity, or teratogenicity
F5: A variable factor that may be applied if the no-effect level was not established
While the calculation in the EMA Guideline differed slightly from the one in Risk-MaPP, the guideline stated that the “PDE and ADE are essentially synonymous.”
The EMA Guideline contains approximately four pages of guidance with references to ICH Q3C (R4) and VICH GL 18 guidelines for further information on the choice of adjustment factors F1 and F4.
The "ADE Supplement"
In 2015, toxicologists from several pharmaceutical organizations and consulting companies met to provide technical details on the various aspects of developing an ADE, which was again considered synonymous with the PDE. The goal of this meeting was to discuss the scientific underpinnings and the technical details of deriving an HBEL and publish them as a reference for the industry. Ten articles were written and published together in Regulatory Toxicology and Pharmacology in 2016 as the "ADE Supplement."11-20 Topics covered by these articles included deriving the point of departure (PoD), selection of adjustment factors, toxicokinetic and toxicodynamic considerations, special endpoints, and product-specific considerations.
Risk-MaPP (Second Edition)
Also in 2015, a team was again formed to adjust Risk-MaPP-V1 to reflect recent changes in regulations, in particular those presented in the EMA Guideline (described above). Risk-MaPP (Version 2) was released in 2017 and included a slightly altered calculation of the ADE value as shown in Equation 3. Previously, version 1 required a NOAEL as the starting point for the calculation, which is not always available. Additionally, other toxicological and clinical data are at times more suitable and potentially superior. Therefore, in Risk-MaPP (Second Edition), the NOAEL was replaced with the PoD and the term Uncertainty Factor (UF) was changed to Adjustment Factor (AF) as depicted in Equation 3.
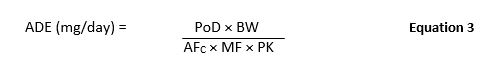
Where:
ADE = Acceptable Daily Exposure (mg/day)
PoD = Point-of-Departure (e.g., NOAEL, LOAEL) (mg/kg/day)
BW = Body Weight (kg)
AFC = Composite Adjustment Factor
MF = Modifying Factor
PK = Pharmacokinetic Adjustment(s)
The second edition of Risk-MaPP provided approximately seven and a half pages of instructions, which also included a table comparing the Adjustment Factors presented in the EMA Guidance with those proposed in the first edition of Risk-MaPP.
ASTM E3219
A potential obstacle to wider adoption of HBEL was the fact that ISPE is not a recognized consensus standard developing organization, therefore limiting the potential for a regulatory authority to adopt the procedures laid out in Risk-MaPP. In addition to this, although industry had the Risk-MaPP and EMA HBEL guidance documents, questions remained on how to correctly utilize the guidelines, and many industry professionals called for further harmonization and systematization of HBEL methodology.
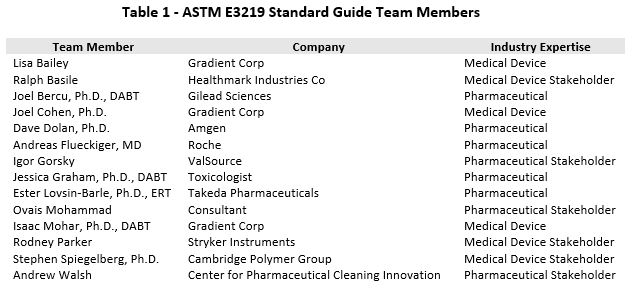
To address these gaps, in July of 2017 a team consisting of toxicologists, along with a medical doctor who had extensive pharmaceutical industry preclinical and clinical experience in the derivation and application of HBELs, was formed to develop an HBEL consensus standard. The team also included several stakeholders from the pharmaceutical and the medical device industries (Table 1).
ASTM International was selected as the organization to develop this standard, as ASTM International is one of the few global consensus standard developing organizations that satisfies the requirements of the U.S. National Technology Transfer and Advancement Act of 1995 so that the FDA may recognize and adopt ASTM standards. The ASTM E3219 team used many of the concepts formulated in the ADE Supplement to provide technical advice on deriving an HBEL to develop this new standard. This new standard should serve as an official reference for HBEL derivations and seeks to harmonize the various approaches and nomenclature being used throughout the industry.
In July of 2018, the draft ASTM standard was completed and balloted in the E55 Committee on Pharmaceutical Standards. After addressing the initial comments received from industry, the standard was re-balloted and approved in December 2019. The new ASTM E3219 standard contains extensive guidance to assist in the derivation of HBELs, is 30 pages in length, and includes example HBEL calculations as well as a template for documenting HBELs (e.g., HBEL monographs). The ASTM HBEL standard was officially published in May 2020 and is available on the ASTM website (https://www.astm.org/Standards/E3219.htm).
Scope Of ASTM E3219
The primary scope of E3219 is to ensure the safety of human patients exposed to residual active substances and intermediates through medicinal products. However, this standard can also be applied to veterinary medicinal products as well as other substances that potentially could leave undesirable residue levels.
E3219 describes the scientific procedures for evaluating all data concerning an active pharmaceutical ingredient (API) and then selecting a point of departure (PoD) in order to inform the derivation of an HBEL for that API. This HBEL can be used in the quality risk management of cross contamination during the manufacture of different products in the same manufacturing facilities. The principles in this guide may be applied during the development and commercial manufacturing of small or large molecular weight medicines as well as isolated pharmaceutical intermediates. This guide should be used for evaluating the hazards of a compound and subsequently calculating and documenting an HBEL, when required, not only for APIs (including biologics) but also for intermediates, cleaning agents, excipients, and other chemicals (i.e., reagents, manufacturing residues, etc.) used for cleaning validation and verification. This guide can be used independently or in conjunction with other ASTM standards, such as E3106.2
Qualified Expert
The establishment of an HBEL is a process that requires expertise and needs to be done by a qualified expert and (if possible) should be peer reviewed by relevant subject matter experts. ASTM E3219 defines a "qualified expert" as an:
"individual with specific education and training in toxicology/pharmacology/ pharmacotherapy and risk assessment methods that can apply the principles of toxicology to deriving an HBEL"
Both the EMA and the Pharmaceutical Inspectorate Cooperation Scheme (PIC/S) in their answer to Q&A #4 on Health Based Exposure Limits21, 22 state:
"Health-Based Exposure Limits should be determined by a person who has adequate expertise and experience in toxicology/pharmacology, familiarity with pharmaceuticals as well as experience in the determination of health-based exposure limits such as Occupational Exposure Levels (OEL) or Permitted Daily Exposure (PDE)".
Also, regarding the use of outside toxicological consultants, the EMA and PIC/S go on to state:
"Where experts are contracted to provide the HBEL, contractual agreements in compliance with Chapter 7 requirements should be in place prior to work being conducted. It is not considered acceptable for manufacturers to ‘purchase’ HBEL assessments without recording an assessment of the suitability of the provider (including the specific technical expert) as a qualified contractor".
The assessment of an individual's qualifications to derive HBELs should include:
- Curriculum vitae (CV) should be available on request that demonstrates the educational background (for example, toxicology, pharmacology, medicine, or other health-related disciplines)
- Years of experience in their field
- Years of experience deriving HBELs
The following professional information may also be useful to include but is optional.
- Certifications such as the Diplomate of the American Board of Toxicology (DABT) or European Registered Toxicologist (ERT)
- Publications related to the field.
The possession of certifications and/or publications does not directly demonstrate competence in the derivation of HBELs. However, certification registries typically require an academic degree in a relevant subject, basic knowledge of the major areas of toxicology, at least five years of relevant toxicological experience, suitability for registration (for example, by published works, reports, or assessments), and current professional engagement in the practice of toxicology. Therefore, the possession of a certification does support acceptance of the qualification of an individual.
While all of the above are not required for a “qualified expert,” appropriate documentation of these areas demonstrates the possession of expertise necessary to work in this area.
ASTM E3219 HBEL Derivation Process
The procedure proposed in ASTM E3219 for determining an HBEL is based on the methodology for establishing the PDE, as described in the EMA guideline, and the ADE value, as described in ISPE’s Risk-MaPP, as well as principles outlined in the scientific literature (see ASTM E3219 references for an exhaustive list). Figure 1 shows a flow diagram of this process.
In E3219, the calculation of the HBEL is conveyed as shown in Equation 4. Note that Equation 4 integrates previous HBEL calculation approaches proposed by the EMA and ISPE, among others.

Where:
PoD = point of departure
FT = composite adjustment factor
PK-AF = Accumulation factor
α = Bioavailability for the route of exposure

Figure 1: ASTM E3219 Example of Process for Final Selection of an HBEL (Reprinted from ASTM E3219-20 Standard Guide the Derivation of Health Based Exposure Limits (HBELs), copyright ASTM International, 100 Barr Harbor Drive, West Conshohocken, PA 19428, USA, www.astm.org.)
Hazard Identification And Characterization
The first step in the calculation of an HBEL is the identification and characterization of the compound’s hazards. This process identifies mechanisms by which agents exert their toxic effects and the associated dose, route, duration, and timing of exposure as well as an assessment of their relevance, reliability, and adequacy for further assessment for the target population. The evaluation of all substance-specific information should result in a comprehensive characterization of the hazards and understanding of the safety profile of the compound.
Identification of the critical effect(s)
The purpose of this step is to identify the biologically significant effect most likely to be relevant for the target population (patients) and target route of exposure (oral, parenteral, other). The “critical effect” has been defined as the “most sensitive adverse effect that is considered relevant to humans” or the “first clinically significant adverse effect that is observed as the dose increases” and “the first adverse effect, or its known precursor that occurs in the increasing dose/concentration scale.” In the case where nonclinical data informs the critical effect, it is necessary to ensure the critical effect is clinically relevant. To evaluate the clinical relevance of an adverse effect, the similarity of effects between animal species and humans and demonstration of homology between the animal model and humans must be evaluated.
Determination of the point of departure (PoD)
The PoD determination builds upon the data collection, dose-response assessment, and identification of the critical effect. The PoD is the highest dose (e.g., mg/kg or mg/person) at which the critical effect is not observed/expected or the lowest dose at which the critical effect is observed/expected. In determining the PoD, all relevant endpoints, including nonclinical and clinical data, must be evaluated. Ideally, the PoD is based upon the No-Observed-Adverse-Effect Level (NOAEL) or the No-Observed-Effect-Level (NOEL) of the most sensitive and/or relevant species for the critical effect(s). When a NOAEL or NOEL is not available, the Lowest-Observed-Effect-Level (LOEL) or the Lowest-Observed-Adverse-Effect-Level (LOAEL) can be used as a PoD. The PoD is then adjusted to account for uncertainties and pharmacokinetic properties to derive an appropriate HBEL relevant for human exposure.
Application Of Adjustment Factors
Once the PoD is identified, adjustment factors (AFs) are applied to adjust for uncertainty and variability in the various parameters measured in the critical study compared to effects that may occur in the target population (the population for which the HBEL is being established to protect).The AFs address the various uncertainties, allowing for extrapolation to a reliable and robust NOAEL in humans to be used for cleaning validation limit calculations.
Pharmacokinetic Adjustments
The E3219 describes the calculation and application of the following pharmacokinetic factors:
- Absorption Factor (α, PK-ABS)
- Accumulation Factor (PK-AF)
The absorption factor is used to extrapolate to alternative routes of administration. For example, a parenteral HBEL value can be derived from oral data, when considering potential for oral bioavailability.
The accumulation factor is also important for deriving an HBEL for an intermittently administered drug. The HBEL assumes daily dosing for a lifetime, and thus the dose of the intermittently administered drug may be lowered to reflect this.
Adjustment Factors (AFs)
The E3219 provides detailed guidance on the following adjustment factors:
- Interspecies extrapolation (F1; UFA)
- Interindividual variability; Intraspecies variability; Human variability (F2; UFH)
- Chemical Specific Adjustment Factor (CSAF)
- Sensitive Subpopulations
- Exposure length AF; Subchronic to Chronic (F3; UFS)
- Severity of effect; Severity (F4)
- LOAEL-to-NOAEL extrapolation (F5;UFL)
- Database completeness (UFD) or Modifying Factor (MF)
- Incomplete Datasets with a High Level of Uncertainty
- Routes of exposure
- Subsequent Product HBEL
The intent here is not to provide an exhaustive list of AFs that toxicologists must provide but tools to critically extrapolate the PoD to an HBEL. The overall goal is to obtain science-based AFs using compound-specific data when possible.
Incomplete datasets with high level of uncertainty
Investigational drugs are typically limited in nonclinical and clinical data such that a data-derived HBEL could not be developed given the irreducible amount of uncertainty (e.g., a composite assessment factor ³5000).17 In addition, compounds such as isolated intermediates will not be extensively safety tested. This section provides approaches for conservatively setting HBELs even when limited data are available.
Route of exposure
Typically, two routes of exposure for HBELs are derived by default, oral and intravenous. The intravenous HBEL can be used for other parenteral routes of administration such as subcutaneous, intramuscular, or intradermal. If an HBEL is required for alternative routes of administration such as ocular, dermal, or inhalation, the sections point to resources that can be used to derive HBELs for these routes. An example of an HBEL for a topical ocular product is in Figure 2.
Subsequent product HBEL
The HBEL assumes that drug substance A could be a residual in any drug substance B. However, on a case-by-case basis, the HBEL for drug substance A could be refined by considering the route of administration, dosing schedule, patient population, or dosing duration of drug substance B.
Special Endpoints
E3219 contains example HBEL calculations and also provides guidance on the following special endpoints:
- Sensitization, including Facility Dedication for Highly Sensitizing Materials
- Genotoxicity (Mutagenicity)
- Reproductive / Developmental Toxicity
- Cytotoxicity
- Potent Molecules (e.g., ADCs)
- Non-human targets: anti-parasitics, antivirals, antibiotics
- Vaccines, Oncolytic Viruses, Gene Therapy
- Over the Counter (OTC) Drugs
- Starting Materials and Intermediates
- Other Chemicals
- Medical Device Considerations
Example HBEL Calculations
Figure 2 shows the derivation of two compounds (Timolol and Ketotifen) with their critical effects, points of departure, and adjustment factors.

Figure 2: Example HBEL Derivation for an Ocular Indication (Reprinted from ASTM E3219-20 Standard Guide the Derivation of Health Based Exposure Limits (HBELs),23 copyright ASTM International, 100 Barr Harbor Drive, West Conshohocken, PA 19428, USA, www.astm.org.)
Documentation For HBELs
Proper documentation of derived HBELs is essential to enable the understanding that the limit is protective for the appropriate population. A description of the necessary information utilized to derive and document the rationale for an HBEL is included in an HBEL monograph. The purpose of an HBEL monograph is to provide an effective communication tool to share with stakeholders, document the scientific principles underlying the HBEL derivation, and enable the inspection of the HBEL documentation by the auditors and regulators. An example template for an HBEL monograph is available in E3219.
Conclusion
This article provides an overview of the new ASTM E3219 Standard Guide For Derivation of HBELs so readers can understand the value and use of the standard as well as the basis for its creation. The authors are confident that the use of HBELs in the quality risk assessment process will reveal significant advantages in most risk management situations.
We hope readers understand the importance of using science in the derivation of HBELs and the necessity of using qualified experts in the development of HBEL monographs. In consideration of the current shortage of expertise and experience, the authors are planning workshops and training courses.
Peer Review
The authors wish to thank Sarra Boujelben, Gabriela Cruz, Ph.D., Ioana Gheorghiev, M.D., Mallory DeGennaro, Parth Desai, Laurence O'Leary, Tri Nguyen Chanh ,Miquel Romero Obon, Prakash Patel, Siegfried Schmitt Ph.D., Stephen Spiegelberg, Ph.D., and Joel Young for reviewing this article and for providing insightful comments and helpful suggestions.
References:
- American Society for Testing and Materials E3219-20 "Standard Guide for Derivation of Health Based Exposure Limits (HBELs)" www.astm.org.
- American Society for Testing and Materials E3106-18e1 "Standard Guide for Science Based and Risk Based Cleaning Process Development and Validation " www.astm.org.
- Andrew Walsh, Thomas Altmann, Joel Bercu, Ph.D., Alfredo Canhoto, Ph.D., David G. Dolan Ph.D., Andreas Flueckiger, M.D., Igor Gorsky, Jessica Graham, Ph.D., Ester Lovsin Barle, Ph.D., Ovais Mohammad, Mariann Neverovitch and Osamu Shirokizawa, "Introduction to Science Based and Risk Based Cleaning Validation and the ASTM E3106 and E3219 Standard Guides". Pharmaceutical Online, May 2020
- Andrew Walsh, Thomas Altmann, Joel Bercu, Ph.D., Alfredo Canhoto, Ph.D., David G. Dolan Ph.D., Andreas Flueckiger, M.D., Igor Gorsky, Jessica Graham, Ph.D., Ester Lovsin Barle, Ph.D., Ovais Mohammad, Mariann Neverovitch and Osamu Shirokizawa "Introduction to the ASTM E3106 "Standard Guide to Science Based and Risk Based Cleaning Process Development and Validation" Pharmaceutical Online, June 2020
- United States vs. Barr Laboratories, Inc. Civil Action No. 92-1744, U.S. District Court for the District of New Jersey: 812 F. Supp. 458. 1993 US Dist. Lexis 1932; 4 February 1993, as amended 30 March 1993.
- Current Good Manufacturing Practice: Proposed Amendment of Certain Requirements for Finished Pharmaceuticals. Federal Register / Vol. 61 No. 87 / Friday, May 3, 1996 / Proposed Rules
- ISPE. (2010). Baseline® Pharmaceutical Engineering Guide: Risk-Based Manufacture of Pharmaceutical Products: A Guide to Managing Risks Associated with Cross-Contamination (First ed. Vol. 7). Tampa, FL: ISPE.
- Code of Federal Regulations Title 21 Part 211 - Current Good Manufacturing Practice for Finished Pharmaceuticals
- Fourman, G., and Mullen, M., “Determining Cleaning Validation Acceptance Limits for Pharmaceutical Manufacturing Operations,” Pharmaceutical Technology, April 1993. Vol. 17 (No. 4): 54-60.
- EMA. (2014). Guideline on setting health based exposure limits for use in risk identification in the manufacture of different medicinal products in shared facilities. London, UK: European Medicines Agency, EMA/CHMP/ CVMP/ SWP/169430/2012, 20 November 2014.
- Bercu, J. P., Morinello, E. J., Sehner, C., Shipp, B. K., & Weideman, P. A. (2016). Point of departure (PoD) selection for the derivation of acceptable daily exposures (ADEs) for active pharmaceutical ingredients (APIs). Regul Toxicol Pharmacol, 79 Suppl 1, S48-56.
- Faria, E. C., Bercu, J. P., Dolan, D. G., Morinello, E. J., Pecquet, A. M., Seaman, C., Sehner, C., & Weideman, P. A. (2016). Using default methodologies to derive an acceptable daily exposure (ADE). Regul Toxicol Pharmacol, 79 Suppl 1, S28-38.
- Gould, J., Callis, C. M., Dolan, D. G., Stanard, B., & Weideman, P. A. (2016). Special endpoint and product specific considerations in pharmaceutical acceptable daily exposure derivation. Regul Toxicol Pharmacol, 79(Suppl 1), S79-S93.
- Hayes, E. P., Jolly, R. A., Faria, E. C., Barle, E. L., Bercu, J. P., Molnar, L. R., Naumann, B. D., Olson, M. J., Pecquet, A. M., Sandhu, R., Shipp, B. K., Sussman, R. G., & Weideman, P. A. (2016). A harmonization effort for acceptable daily exposure application to pharmaceutical manufacturing - Operational considerations. Regul Toxicol Pharmacol, 79 Suppl 1, S39-47.
- Olson, M. J., Faria, E. C., Hayes, E. P., Jolly, R. A., Barle, E. L., Molnar, L. R., Naumann, B. D., Pecquet, A. M., Shipp, B. K., Sussman, R. G., & Weideman, P. A. (2016). Issues and approaches for ensuring effective communication on acceptable daily exposure (ADE) values applied to pharmaceutical cleaning. Regul Toxicol Pharmacol, 79 Suppl 1, S19-27.
- Reichard, J. F., Maier, M. A., Naumann, B. D., Pecquet, A. M., Pfister, T., Sandhu, R., Sargent, E. V., Streeter, A. J., & Weideman, P. A. (2016). Toxicokinetic and toxicodynamic considerations when deriving health-based exposure limits for pharmaceuticals. Regul Toxicol Pharmacol, 79 Suppl 1, S67-78.
- Sargent, E. V., Flueckiger, A., Barle, E. L., Luo, W., Molnar, L. R., Sandhu, R., & Weideman, P. A. (2016). The regulatory framework for preventing cross-contamination of pharmaceutical products: History and considerations for the future. Regul Toxicol Pharmacol, 79 Suppl 1, S3-S10.
- Sussman, R. G., Naumann, B. D., Pfister, T., Sehner, C., Seaman, C., & Weideman, P. A. (2016). A harmonization effort for acceptable daily exposure derivation - Considerations for application of adjustment factors. Regul Toxicol Pharmacol, 79 Suppl 1, S57-66.
- Sussman, R. G., Schatz, A. R., Kimmel, T. A., Ader, A., Naumann, B. D., & Weideman, P. A. (2016). Identifying and assessing highly hazardous drugs within quality risk management programs. Regul Toxicol Pharmacol, 79 Suppl 1, S11-18.
- Weideman, P. A., Pecquet, A. M., & Maier, M. A. (2016). Harmonization efforts for deriving health-based exposure limits in the pharmaceutical industry - Advancing the current science and practice. Regul Toxicol Pharmacol, 79 Suppl 1, S1-2.
- European Medicines Agency: Questions and answers on implementation of risk-based prevention of cross-contamination in production and “Guideline on setting health-based exposure limits for use in risk identification in the manufacture of different medicinal products in shared facilities," 19 April 2018 EMA/CHMP/CVMP/SWP/246844/2018
- PIC/S. (2020). Questions and Answers on Implementation of Risk-Based Prevention of Cross-Contamination in Production and ‘Guideline on Setting Health-Based Exposure Limits for Use in Risk Identification in the Manufacture of Different Medicinal Products in Shared Facilities’ (PI 053-1).
- Lovsin Barle E., Glogovac M., Gromek K., Winkler GC, Milton M., Bizec J., Newton R. PDE method development for topical ocular Products Toxicology Letters 258S (2016) S62–S324.