
ICAT -- A New Method for Quantitative Proteome Analysis

Two-dimensional gel electrophoresis, a commonly used tool in proteomics, is rather cumbersome, difficult to automate, and works better with some kinds of proteins than with others. Membrane proteins, for example, or very basic or very acidic proteins aren't well resolved with most two-dimensional gel systems. In contrast, ICAT shows no such bias for particular proteins. In addition, it permits concurrent analysis of more than one sample, allowing the precise measurement of changes in expression level with even rare proteins.
In ICAT, Ruedi Aebersold and his colleagues have combined an existing technique—mass spectrometry—with radiolabeled tags to develop a rapid, simple, and quantitative protein detection method. ICAT reagents consist of three elements—a specific chemical reactivity towards sulfhydryl groups, a linker bearing an isotopic label like deuterium, and an affinity tag, such as biotin.
Preparing samples for analysis requires four steps:
- Cysteine side chains from one sample are deriviatized with the labeled linker, from a second sample with unlabeled linker.
- The two samples are mixed and subjected to limited proteolysis.
- The biotin-tagged peptides are captured by affinity chromatography.
- The resulting peptides are analyzed by microcapillary liquid chromatography coupled to automated mass spectroscopy, which provides information on the quantity and identity of the peptides.
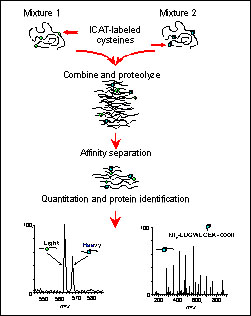
The schematic illustrates the method in a simplified form, showing the labeling of a single protein at its cysteine residues, and the subsequent quantitation and identification of the resulting peptides. This is essentially what happens with a complex protein mixture, but on a grander scale. The mass spectrometer (MS) operates in two stages (i.e. MS mode and MS/MS or tandem MS mode). The MS mode measures peptide ions from the ICAT-labeled peptide pairs. This ratio reveals the ratio of the proteins in the original mixture. The second stage of MS is for sequencing. A peptide ion is selected and fragmented and then computer matched to a protein database. This illustrates how relative quantitation of protein levels and sequence identification can be done in a single analysis for complex protein mixtures.

The second figure is a tandem mass spectrum showing how the peptide is "sequenced" in a mass spectrometer. In the example shown, the best-matched computer-identified sequence from the yeast database was BMH1 with a modified cysteinyl residue.
To validate the ICAT approach, Aebersold's team studied protein levels in yeast grown in either galactose- or ethanol-containing media. When they analyzed the fingerprints from the mass spectrometer, they correctly identified and quantitated proteins known to be induced or derepressed under growth in galactose or ethanol.
Aebersold believes the ICAT approach should be broadly applicable for the quantitative measurement of protein expression in cells in many different stages of development or disease, with broad relevance to pharmaceutical and basic biology researchers.
For more information: Ruedi Aebersold, University of Washington, Department of Molecular Biotechnology, Box 35770, Seattle, WA 98195-7730. Tel: 206-221-4196. Fax: 206-685-7301. Email: ruedi@u.washington.edu.