
How To Minimize The Impact Of Stability Testing On Gene Therapy Batch Yield
By BioPhorum

It is widely recognized that gene therapy manufacturing processes result in low yields, particularly in early product development stages. Gene therapy products, however, are largely subject to many of the same regulatory requirements and expectations as other large molecule biopharmaceuticals that are produced at significantly larger scales. Often, if gene therapy manufacturers adhered to common paradigms for stability studies, the outcome would be little, if any, remaining product for patients or studies to support investigational new drug applications.
This article outlines strategies for reducing the volumes required for stability studies, with the goal of conserving product for patients, while remaining compliant and delivering data on critical quality attributes across the shelf life of gene therapy products. The recommendations include considerations for both drug substance (DS) and drug product (DP).
The scope of this article is limited to viral vector gene therapies; however, the concepts discussed may be applied to other modalities within cell and gene therapies (CGT), such as ex vivo genetically modified human cells. The need for specific recommendations for CGT products has been identified by observing that stability guidance (e.g., ICH Q11 and ICH Q52) is mainly designed for large-scale biologic/small molecule products and is not fully applicable to the unique specificities of gene therapy products. The recommendations focus on strategies for routine stability studies.
To address these challenges collaboratively, we conducted a survey to identify the areas of greatest impact on batch yield. The survey identified the highest-ranking items with an adverse impact on batch yield as bioburden and sterility,1 container closure integrity testing and reserve samples,2 and stability. To further understand the impact of stability on batch yield, a follow-up survey was conducted in Q2 2021 to assess the regulatory guidance applied and approaches taken by member companies in executing stability studies for CGT products. The key results from this survey are included in this article.
Figures 1 and 2 show the wide variety of stability studies conducted by the member companies and their impact on batch yield. The most frequently performed studies, and those with the greatest impact on batch yield, are those required to support the establishment of routine shelf life; therefore, the scope of further review was focused on routine stability studies.
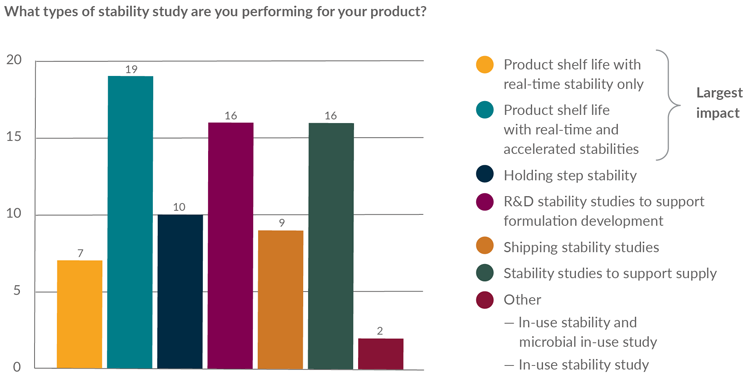
Figure 1: Stability survey question—types of stability studies performed
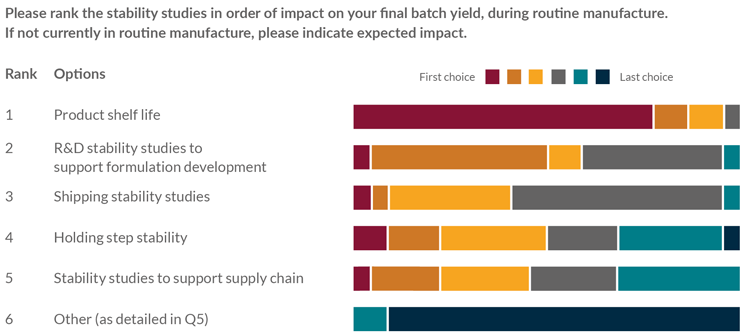
Figure 2: Stability survey question—impact on batch yield
Figure 3 shows a sample of strategies that surveyed members use for the conservation of material during the design and execution of stability studies. The most common of these focused on adapting the design of the study to streamline the number of time points, limiting stability studies to the intended storage condition of the product, and/or eliminating DS stability studies. Approaches leveraging data from prior experience with other products were also mentioned.
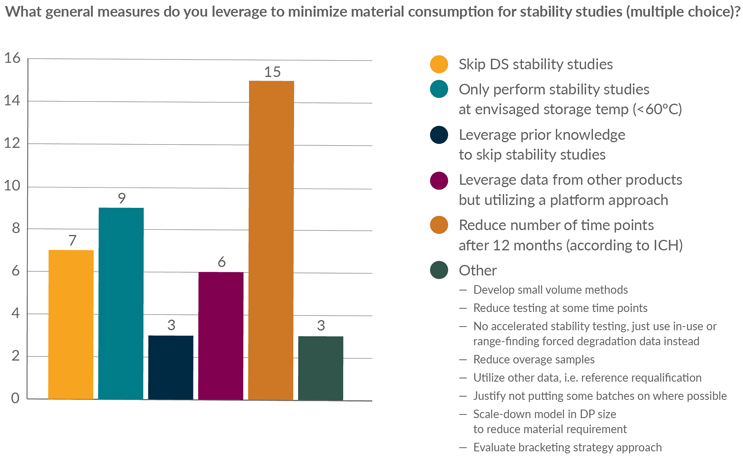
Figure 3: Stability survey question—strategies to minimize the impact on batch yield
These survey results highlight that member companies agree that applying a one-size-fits-all approach to the execution of stability studies for CGT products is not feasible. Alternate solutions are required to minimize the impact on batch yield; however, specific recommendations vary based on product type. To provide specific strategies that member companies could consider for implementation, we defined a mock viral vector product and used it to identify the strategies for the conservation of material.
Mock Product And Testing Assumptions
To avoid getting lost in generic considerations that might not be applicable to CGT products, a stability assessment was performed based on a defined case study.
Consequently, efforts were focused on defining the quality attributes and manufacturing process characteristics of a representative in vivo gene therapy product. As illustrated in Table 1, the recombinant adeno-associated virus (rAAV) mock product would be produced at 200-L scale in HEK293 suspension cells using a three-plasmid transient transfection process. In brief, one batch of DS would represent 250 mL of viral suspension at 4x1013 vg/mL (a total of 1x1016 vg) stored in a 500-mL bag. From this DS batch, one DP batch (500 vials) would be produced with a targeted dose of 1x1013 vg in 2-mL vials filled with a total volume of 1 mL (1x1013 vg/mL). Both DS and DP would be frozen and stored at < minus 60 degrees C.
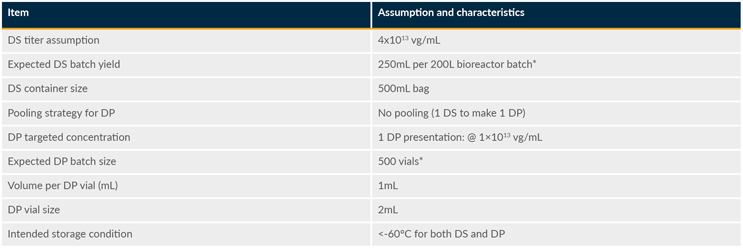
Table 1: rAAV mock product assumptions and characteristics
Having defined the characteristics of the product that would be placed on stability, the stability-indicating analytical package and the typical volumes of DS and DP per test and time point were defined to support the baseline of the stability program.
Fifteen quality attributes were deemed stability-indicating and covered strength, potency, purity, impurity, and safety aspects. The analytical package and associated sample volumes, representing 26 mL of DS and 41 vials of DP per time point, were used as a baseline for the mock stability program described in the next section.
Mock Stability Program
Based on the mock product assumptions, characteristics, and 15 selected quality attributes, we defined a baseline stability program design and created a set of recommendations based on specific CGT considerations and challenges.
Baseline (Worst-Case) Stability Program And Guidance
The baseline strategies for DS and DP stability are outlined in Tables 2 and 3. The baseline strategy includes a comprehensive analytical method testing panel and includes volume requirements needed at each stability time point. The baseline strategy is also based on the recommended stability time points per ICH guidelines and extends to 36 months for both DS and DP. The baseline strategy does not incorporate any strategies for conserving materials to demonstrate a worst-case scenario of material use required by the stability program and, therefore, highlights the full “stability tax” on batch yield.
The baseline stability program for the mock product DS stability is outlined in Table 2. Based on the analytical test method panel and volume needed to perform each assay presented in the table, the DS stability container (a scale-down bag representative of the DS storage container) will need a minimum fill volume of 5.85 mL (inclusive of overage) to complete all required testing at each of the given stability time points. For the intended storage condition (< minus 60 degrees C), eight time points are planned, monitoring stability for up to 36 months. Therefore, the required DS volume needed for the stability program, at just the intended storage condition, is 58.5 mL (inclusive of two extra stability containers as backup).
Table 2: Example of the baseline drug substance stability program.
Key: AUC—analytical ultracentrifugation, CBA—cell-based assay, DLS—dynamic light scattering,
dPCR—digital PCR (polymerase chain reaction), ELISA—enzyme-linked immunosorbent assay,
LAL—limulus amoebocyte lysate, SDS-CGE—capillary SDS gel electrophoresis,
SDS-PAGE—sodium dodecyl sulfate-polyacrylamide gel electrophoresis,
TCID50—50% tissue culture infectious dose
The baseline stability program for the mock product DP stability is outlined in Table 3. Based on the analytical test method panel and number of containers required to perform each assay, as well as the selection of test methods at each stability time point presented in the table, a minimum of 232 DP vials will be required to perform the stability study as planned at just the intended storage condition (< minus 60 degrees C), monitoring DP stability for up to 36 months.
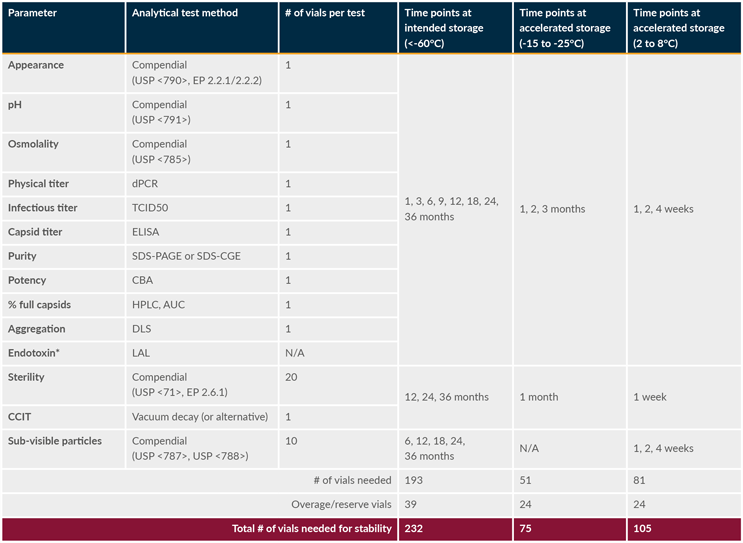
Table 3: Example of baseline DP stability program
Key: AUC—analytical ultracentrifugation, CBA—cell-based assay,
CCIT—container closure integrity testing, DLS—dynamic light scattering,
dPCR—digital PCR (polymerase chain reaction), ELISA—enzyme-linked immunosorbent assay,
HPLC—high-performance liquid chromatography, LAL—limulus amoebocyte lysate,
SDS-CGE—capillary SDS gel electrophoresis,
SDS-PAGE—sodium dodecyl sulfate-polyacrylamide gel electrophoresis,
TCID50—50% tissue culture infectious dose
Unique CGT Considerations, Challenges, And Recommendations/Strategies
As previously stated, for CGT products, the manufactured DS and DP batch sizes are typically small, and the current release and stability material requirements are, therefore, prohibitively high, requiring large amounts of sample volume and depleting a significant portion of the batch. Alternative strategies are needed to optimize stability protocols and reduce the impact on batch yield. Several strategies were identified that offer the potential to optimize the baseline stability protocols and support efforts to conserve product and reduce the material requirements to perform DS and DP stability. Optimization strategies have been separated into categories, including sample volumes, analytical methods, study protocol, and others.
Sample Volumes
Reducing the sample volume required for analytical testing is advantageous for conserving DS and DP material needed for stability. Strategies have been identified as implementation examples of how to reduce sample volume requirements, and they include:
- Where feasible, multiple assays should share the same sample container instead of providing individual containers for each test (e.g., appearance, pH, osmolality). Sharing a single container enables a reduction in overage and the unnecessary use of additional containers when a single container is feasible.
- Preferentially select contract labs for outsourced testing with the capability to perform multiple assays, where shared containers can be used.
- The overage required for the stability study can be determined as a percentage of the total number of containers required for the duration of the study, as opposed to accounting for extra containers for each assay/time point.
Analytical Methods
The selection of analytical methods to include in the stability protocol has a direct impact on the material requirements. Strategies have been identified to aid in the selection of analytical test methods and highlight opportunities to conserve material, including:
- Methods included in the stability protocol should be limited to stability-indicating assays.
- Where orthogonal test methods are used for release testing, for stability testing, consider the use of the test method with the lesser volume requirements (e.g., high-performance liquid chromatography vs. analytical ultracentrifugation for % full capsids).
- Where available, leverage the use of microprobes or smaller volume techniques (e.g., the use of a small volume capable osmometer for osmolality testing).
Study Protocol
Opportunities to optimize the stability study protocol were also identified as a means to reduce the stability study material requirements. These strategies have been identified as:
- The length of the DS stability study should be limited to the storage time required prior to the DP fill rather than set at an arbitrary number of years.
- Attributes unlikely to change in the short term can be tested annually instead of at every time point. This can reduce the material requirements at the intermediate protocol time points.
- Where applicable, reduce the number of time points and temperatures being evaluated.
Other Strategies
Strategies and recommendations were identified as additional opportunities to reduce the material needed for stability. These include:
- Consolidate the testing sites (ideally using all in-house test methods where feasible) to reduce splitting and sharing vials across different sites. There may be opportunities to use fewer vials in one location where testing is occurring at the same time.
- Review whether dilution is required for the test assay when being performed. If so, there may be an opportunity to provide a smaller test volume of the concentrated sample.
- Perform statistical analysis to justify the use of smaller sample plans since the final batch size is smaller. Also factored into this would be probabilistic considerations of the likelihood of a failure based on historical data, which would impact the overage required.
Conclusion
The potential of CGTs to transform the way we treat and cure disease has led to substantial growth in the development of these new treatment modalities. However, the overall yield of gene therapy manufacturing processes, in terms of both volume of DS and filled DP vials, presents challenges to following the typical stability study approaches used for traditional biopharmaceuticals.
This article is based on a survey of industry participants and was used to identify ways to reduce the impact of routine stability testing on batch yield. Approaches for organizations to consider include (listed in estimated order of impact):
- performing product development studies on representative non-GMP material,
- including expanded storage conditions and time points in research stability studies,
- limiting DS stability time points to one year,
- using container closure integrity testing in place of sterility,
- implementing low-volume methods (e.g., osmolality, pH, titer, % full capsids, and potency), and
- sharing samples and the concurrent scheduling of common analyses at common testing sites.
As demonstrated with the mock rAAV process and associated stability study, product savings of up to 17% of a DS batch and 44% of a DP batch relative to the baseline protocol may be possible.
References
- Minimizing the impact of container closure integrity testing on gene therapy batch yield, BioPhorum, March 2023
- Minimizing the impact of bioburden and sterility testing on gene therapy batch yield, BioPhorum, June 2023
This article summarizes a recent BioPhorum publication on the topic. To read more, including the full case study using the mock product, check out the full paper Minimizing the impact of stability testing on gene therapy batch yield.