
How Do FDA, MHRA, & Russia's Drug Inspectorate Evaluate & Escalate Inspection Findings?

At the FDA/Xavier PharmaLink conference earlier this year, representatives from the Russian Federation State Institute of Drugs and Good Practices, the U.K.’s Medicines and Healthcare products Regulatory Agency (MHRA), and the U.S. FDA shared their top 10 pharma company inspection findings from FY2019, revealing many similarities and a few differences (see related article).
In addition, each also shared its processes for evaluating and escalating inspection findings to determine if regulatory action is needed, which are reviewed here.
Director of Investigations Branch in the division of Pharmaceutical Quality Operations III within FDA’s Office of Regulatory Affairs Lieutenant Commander Jeffrey Meng discussed what he called the “stereotypical regulatory process for a CGMP warning letter” (see Figure 1).

Figure 1
The process begins with an FDA investigator performing an inspection and issuing a 483 to the firm’s management that contains the investigator’s observations. The investigator will complete the Establishment Inspection Report (EIR), with the supporting evidence.
“There is a supervisory review process in which someone like me — a director of investigations branch — will have input into whether the case will proceed or not,” Meng explained. “If the management review within the investigation branch determines that a case should be put forward, it will be sent to a compliance officer for a first-line technical review and legal review. Typically, a director of compliance branch, or DCB, also provides input.”
If the case is deemed significant — and at this point a firm's response is also taken into consideration — the case might be submitted to the appropriate center, such as the Center for Drug Evaluation and Research or the Center for Veterinary Medicine, for example. And there will be a center compliance officer review of the case, as well as decisions made by center management that consider current policy and legal assessments. “If all those things play out properly and there are no mitigating factors, the warning letter might be issued,” Meng said.
MHRA Uses Compliance Management And Inspection Action Group Processes
The U.K.’s MHRA GMDP Senior Inspector and GMDP Operations Manager Graham Carroll explained his agency’s processes for classifying inspection findings and its use of compliance management and Inspection Action Group processes.
Carroll began his explanation using a few inspection finding examples. He started with a case in which an investigation of a deviation did not contain sufficient detail to describe the nature of the deviation, the assessment of the impact on products, or the identification of a root cause. There are also examples in the same inspection of a failure to follow the existing deviation procedure, and there is a repeat of the finding from the previous inspection. “The nature of these observations would be an instant referral to what we call our compliance management team,” he explained.
In another example, the management of out of specification investigations was deficient. Summary reports were not sufficiently comprehensive. The reports had come from a contract lab and were lacking in detail. The manufacturer had not secured additional information or provided a suitable justification for the conclusions drawn. “The second finding is going to be considered significant and would be raised to the compliance management team at a minimum.”
Carroll shared MHRA’s compliance management process overview (Figure 2).
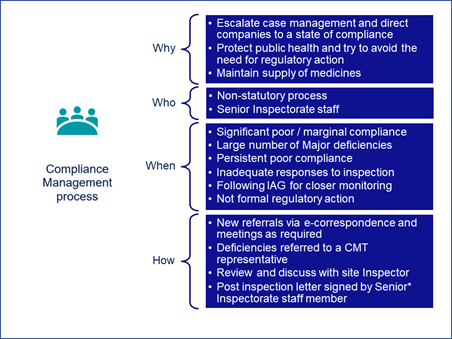
Figure 2
Carroll explained that the process is non-statutory and is managed within the inspectorate by senior and expert inspectors, operations, and unit managers. The team meets monthly or ad hoc as required. “Really the aim of this group is to escalate cases where we can help direct companies to a state of compliance before they are directly putting public health at risk. The aim is to avoid regulatory action and maintain supply of medicines,” Carroll explained.
A different group, the Inspection Action Group (IAG) (Figure 3), “comes into play whenever there is a potential need for us to take regulatory action,” Carroll said. “It is a cross-functional team that encompasses expertise from across the agency and external bodies where needed.”
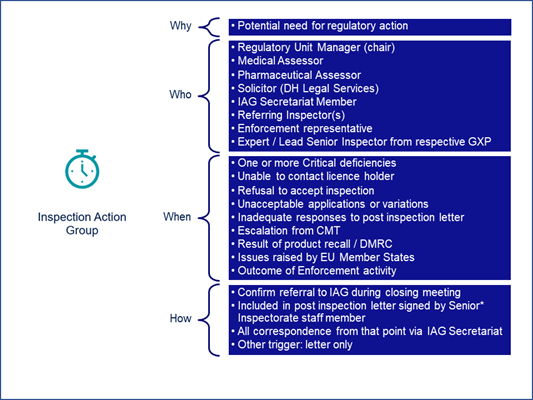
Figure 3
Sites are referred to the IAG typically for one or more critical deficiencies found during an inspection. But referrals can also come from other causes such as refusal to accept an inspection or lack of or poor responses or other quality issues — for example, a large or significant recall.
“After an inspection, sites will be told at the closing meeting that a referral to IAG is likely. Company management will be provided with a link to more information to help them understand the potential outcomes and support their accurate and timely communication with the inspector and the inspection action group,” Carroll said.
That group meets every two weeks routinely but does hold emergency meetings for urgent referrals.
Carroll explained that there are different possible outcomes between U.K. sites and those not in the U.K. due to different licensing arrangements and other factors. The different possible outcomes are listed in Figure 4.
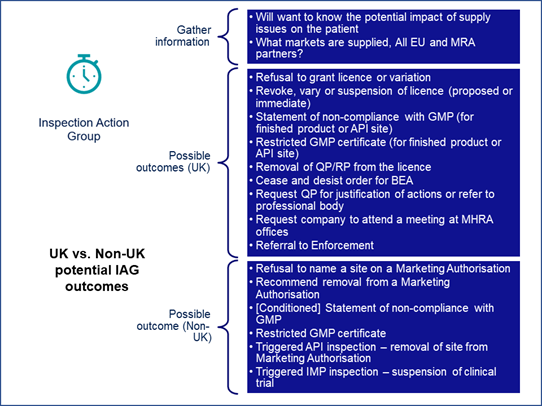
Figure 4
“It's worth bearing in mind that in both cases, the IAG does have the power to prevent supply to the U.K. and EU markets and can also prevent or impact supply to Mutual Recognition Agreement (MRA) partner markets, where we make a recommendation that products coming out of that facility are not safe,” Graham noted.
Russian Federation State Institute of Drugs and Good Practices Process Similar To MHRA’s
Joining the FDA/Xavier PharmaLink conference in 2020 was Deputy Head of Expertise, Department of State Institute of Drugs and Good Practices of the Ministry of Industry and Trade, Lead GMP Inspector in the Russian Federation Ministry of Health Nadezhda Arkhipova.
When asked about the Russian Federation’s system for making decisions on how to deal with inspection findings and potential escalations, Arkhipova said, “Our system is not all that different from the one MHRA has. If we identify critical or major issues, we will provide those to the company in an inspection report. Of course, the company must provide some proof they have made progress on fixing their problems, and then we inspect them again. We do not issue any letters based on our findings in our final inspection report.”
About The Author:
 Jerry is Govzilla’s senior GMP quality expert. He brings 40 years’ experience in the pharma industry, including 31 years at Eli Lilly, where he worked in product development, biosynthetic human insulin manufacturing, and site and corporate quality roles. In corporate quality, he designed and implemented a comprehensive GMP Intelligence process to identify, analyze, and archive pertinent drug GMP regulations, inspection findings, trends, and best practices in the U.S. and internationally. Chapman was founder and chairman of the GMP Intelligence subgroup of the Midwest Discussion Group from 2005 to 2010. He also served as senior editor at International Pharmaceutical Quality for six years; has been an invited speaker at PDA, AAPS, ISPE, and RAPS events; and was a consultant for the animal health and compounding pharmacy industries.
Jerry is Govzilla’s senior GMP quality expert. He brings 40 years’ experience in the pharma industry, including 31 years at Eli Lilly, where he worked in product development, biosynthetic human insulin manufacturing, and site and corporate quality roles. In corporate quality, he designed and implemented a comprehensive GMP Intelligence process to identify, analyze, and archive pertinent drug GMP regulations, inspection findings, trends, and best practices in the U.S. and internationally. Chapman was founder and chairman of the GMP Intelligence subgroup of the Midwest Discussion Group from 2005 to 2010. He also served as senior editor at International Pharmaceutical Quality for six years; has been an invited speaker at PDA, AAPS, ISPE, and RAPS events; and was a consultant for the animal health and compounding pharmacy industries.