
Here's A Thought: What If Gene Editing Didn't Use Bacterial Enzymes?

A conversation with Arun Srivastava, Ph.D., nAAVigen Therapeutics , and Life Science Connect's Executive Editor Jon O'Connell

Here’s a potentially unpopular opinion: enzymes may not be the best tools to edit genes.
Arun Srivastava, Ph.D., a researcher and gene therapy developer at the University of Florida, is exploring a compelling alternative. In light of the groundbreaking approval of Casgevy, a gene therapy based on CRISPR/Cas9 technology, the premise sounds tenuous. While Casgevy is a powerful example, the stock values of all enzyme-based gene editing companies have tanked following vigorous IPOs.
He’s proposing a radically different path forward. Instead of cutting DNA with nucleases like CRISPR/Cas9 or relying on base or prime editors, his team is developing a nuclease-free approach that reactivates dormant genes using synthetic zinc finger DNA-binding peptides. Delivered via AAV6 vectors and expressed under an erythroid-specific promoter, these peptides aim to safely induce fetal hemoglobin expression — offering a potential treatment for sickle cell disease and β-thalassemia without the risks of chromosomal damage or immune response.
We asked Srivastava to explain the science behind this enzyme-free strategy, the safety concerns driving its development, and why he believes it could help restore confidence in the future of gene editing. Here’s what he told us.
Can you start by catching us up on the basics of nuclease-free genome editing? What are the practical parts of your approach? What's being edited if there's no nuclease cutting the DNA?
Srivastava: My colleague and collaborator, Jörg Bungert, Ph.D., professor of biochemistry and molecular biology, here at the University of Florida. has pursued basic research on globin gene regulation for more than three decades. He has developed synthetic zinc finger DNA-binding peptides (ZF-DBD) that function as decoy transcription factors, bind to regulatory sequences in the human globin gene with remarkable specificity, and lead to reactivation of fetal hemoglobin (HbF) expression. This is depicted schematically in Figure 1.
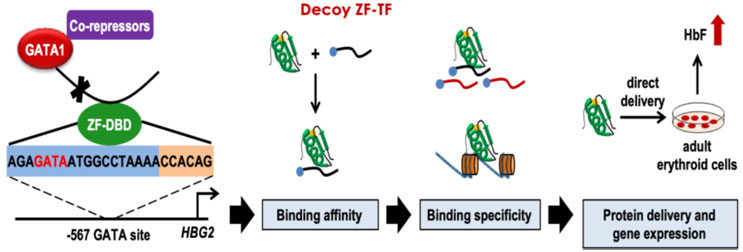
Figure 1: Schematic representation of decoy zinc finger DNA-binding peptide (ZF-DBD)-mediated reactivation of fetal hemoglobin (HbF) expression in human erythroid progenitor cells.
Bungert and his colleagues were able to document a dose-dependent increase in HbF not only in peripheral blood mononuclear cell-derived normal human erythroid cells but also in bone marrow-derived erythroid cells from patients with sickle cell disease (SCD) by simply incubating these cells with decoy ZF-BPD peptides.1 Moreover, these peptides were also shown to significantly inhibit sickling of red blood cells (RBCs) from SCD patients.2 Despite these compelling observations, however, induction of HbF is transient because of the low efficiency of entry of these peptides into cells and their rapid turnover.
A little over a decade ago, work from my laboratory led to the identification of AAV6 as the most efficient vector for transducing primary human hematopoietic stem cells (HSCs).3 Incidentally, AAV6 vectors have also become the vector of gene editing in primary human HSCs. Furthermore, we also documented that expression from a promoter from the human parvovirus B19 (B19V), termed B19p6, was restricted to primary human erythroid cells.4-9
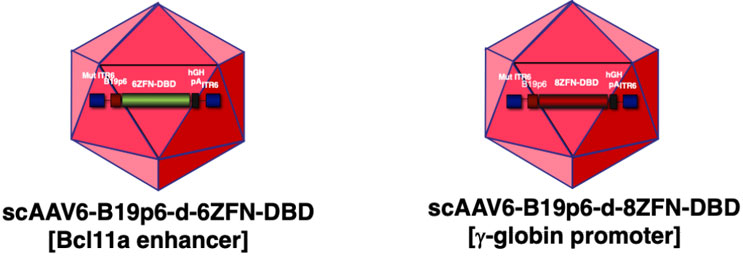
Figure 2: Schematic representation of self-complementary AAV6 vectors containing 6- and 8-decoy ZF-DBD peptide expressing genes under the control of the human erythroid cell-specific B19p6 promoter.
Therefore, we reverse engineered the decoy ZF-DBD peptide amino acid sequence into DNA, placed it under the B19p6 promoter, and packaged it into AAV6 vectors. As a matter of fact, we generated two such vectors, shown schematically in Figure 2: the 6-decoy ZF-DBD peptide expressing gene targeting the Bcl11a enhancer and the 8-decoy ZF-DBD peptide expressing gene targeting the human g-globin gene promoter sequences under the control of the human erythroid cell-specific B19p6 promoter (unpublished data). Thus, sustained and erythroid lineage restricted expression of these peptides promises to be a safe and effective strategy to achieve gene editing.
So, in essence, our approach does not rely on cutting the DNA but rather reactivating the therapeutic gene by specific binding of decoy ZF-DBD transcription factors to the regulatory sequences of human globin genes.
What specific safety concerns with CRISPR/Cas9 and base/prime editors are you trying to avoid with a nuclease-free approach? Are we talking about off-target effects, chromosomal rearrangements, or is it something else?
Srivastava: Although CRISPR/Cas9 targeting the Bcl11a gene in patients with transfusion-dependent β-thalassemia and in patients with SCD has shown clinical efficacy,10 the use of CRISPR/Cas9 has also been shown to led to chromosomal translocation and chromosomal loss in murine hematopoietic stem cells and in primary human T cells.11, 12 Similarly, although base editors and prime editors have shown efficacy in correcting the sickle mutation in mouse models,13, 14 their use has also been shown to be genotoxic in primary human HSCs.15 Thus, the long-term safety of CRISPR/Cas9 and base and prime editors in humans remains to be determined.
Is your approach actually correcting the mutant gene, or are you delivering a functional gene copy that compensates for the mutation?
Srivastava: Our approach involves neither correcting the mutant gene, nor are we delivering a functional gene copy that compensates for the mutation. Instead, we reactivate the endogenous human g-globin gene, which, in turn, should not only ameliorate the symptoms of β-thalassemia but also prevent RBC sickling.
How do you reach integration or stable expression without nuclease activity? Is this episomal, or are you getting genomic integration through AAV's natural mechanisms?
Srivastava: This is an important question, which warrants a detailed discussion. Integration of the recombinant AAV genome has been the subject of some debate. For example, for over a quarter of a century, it has generally been assumed that recombinant AAV vector genomes remain episomal and can form extrachromosomal concatemers,16-18 but those studies were carried out in post-mitotic tissues,19-21 and there was little selective pressure to integrate into the host chromosomal DNA. In HSCs, on the other hand, in the absence of stable integration, the recombinant AAV genomes would be expected to be lost in HSCs. However, HSCs, although largely quiescent, do divide slowly and undergo limited cell divisions.22, 23 Thus, we hypothesized that recombinant AAV DNA might undergo integration into host chromosomal DNA in HSCs.
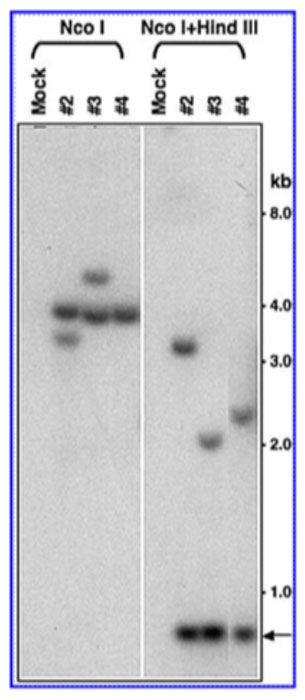
Figure 3: Southern blot analysis for integration of recombinant AAV-b-globin genomes in mouse bone marrow progenitor cells 3 months post-secondary transplantation.
We tested this hypothesis, albeit using mouse HSCs, in two sets of serial stem cell transplantation studies. In the first set, we documented that following ex vivo transduction, the recombinant AAV-human b-globin vectors mediated sustained transgene expression in bone marrow progenitor cells for up to six months, suggesting stable integration of the vector DNA sequences in primary transplant recipient mice. Six months post-primary transplantation, HSCs from primary recipient mice were isolated and transplanted into lethally-irradiated secondary transplant recipient mice. Three months post-secondary post-transplantation, genomic DNA isolated from bone marrow cells were analyzed by Southern blot analysis (Figure 3), which corroborated stable integration of the AAV vector genomes in bone marrow progenitor cells.24
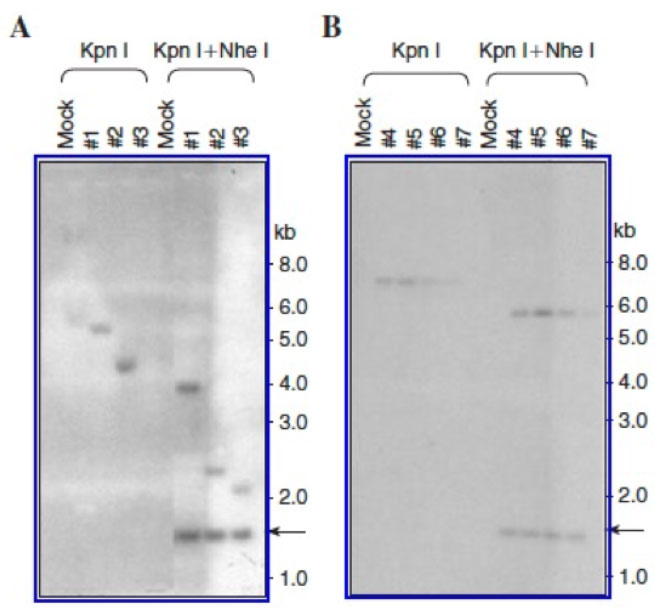
Figure 4: Southern blot analysis for integration of recombinant AAV genomes in mouse bone marrow progenitor cells 6 months post-primary transplantation (A) and 6 months post-secondary transplantation (B).
In the second set, we also documented stable integration of the recombinant AAV-Zeomycin genomes in bone marrow progenitor cells six months post-primary transplantation (Figure 4A). Six months post-primary transplantation, HSCs from one primary recipient mouse were transplanted into four lethally-irradiated secondary transplant recipient mice. Six months post-secondary transplantation, genomic DNA isolated from bone marrow progenitor cells also showed an identical integration pattern of the AAV vector genomes (Figure 4B).25
We subsequently documented stable integration of recombinant AAV genomes in primary human HSCs in long-term cultures (unpublished data). Thus, in contrast to post-mitotic cells, my contention is that in quiescent/slowly dividing HSCs, AAV undergoes genomic integration, leading to erythroid lineage-restricted sustained transgene expression from the B19p6 promoter following differentiation.
Stable integration of AAV genomes leading to long-term transgene expression in hepatocytes has also been documented in canines (up to 10 years)26, and in non-human primates (up to 2 years)27. Although direct evidence of integration of AAV genomes in patients could not be obtained, sustained transgene expression for up to 13 years was recently reported28, which was unlikely from the episomal genomes, which would be expected to be gradually lost because of hepatocyte turnover (~12 months in adults), and thus, probably from the integrated genomes.
Your recent talk highlighted the dismal prospects of traditional Cas-mediated editing companies. Does the enzyme-free approach, in your opinion, help put genetic editing back on the right track?
Srivastava: I should clarify that in my talk, I only referred to the decline in investors’ confidence in all seven gene editing companies, as reflected by the loss of their market cap values over the past five years, in contrast to the steady upward trajectory of the Dow Jones industrial average. Thus, my conclusion was that although the current versions of CRISPR/Cas9, base editors, and prime editors are highly efficient for gene editing in cultured cells in vitro, and in mice and in non-human primates in vivo, these bacteria-derived nucleases and their derivatives may be immunogenic and, thus, not optimal in humans.
It should be noted that in two Phase 3 trials, sponsored by Vertex Pharmaceuticals, in patients with severe sickle disease and transfusion-dependent β-thalassemia, respectively, >90% of the patients, whose HSCs were transduced with lipid nanoparticle (LNP) vectors ex vivo, followed by myeloablation and autologous stem transplantation, benefited significantly.29, 30 Thus, it is unlikely that the Cas9 protein encountered the host immune system. A clinical trial, sponsored by Novartis and Intellia Therapeutics, also involving CRISPR/Cas9-mediated ex vivo editing to disrupt the b-globin gene promoter, was discontinued in 2023, and Editas Medicine discontinued its CRISPR/Cas12a-mediated gene editing program for SCD in 2024 (Mitchell Weiss, 10th Annual Cell and Gene Therapy Symposium, Centre for Stem Cell Research, Christian Medical College, Vellore, India, October 10, 2025).
However, when CRISPR/Cas9 was used in a liver-directed Phase 2 clinical trial for hereditary angioedema, 73% of the patients transfused intravenously with the highest dose of LNPs also benefited for up to week 16, with no serious adverse events,31 but given the immune-tolerance nature of the liver, this is perhaps not unexpected.
In a recent remarkable clinical trial,32 an infant with neonatal-onset carbamoyl-phosphate synthetase 1 (CPS1) deficiency was treated successfully with adenine base editors (ABEs) encapsulated in LNPs infused intravenously at 7 months and at 8 months of age. Thus, it also remains possible that the ABE proteins led to no adverse event, given the immature state of the immune system at that age. It should also be stated that ABEs have been shown to be associated with high off-target activity, leading to genomic rearrangements (Toni Cathomen, 28th Annual Meeting, American Society of Gene and Cell Therapy, New Orleans, LA, USA, May 16, 2025).
It is certainly possible that for the next generations of CRISPR/Cas9 that are being developed,33 or will be generated in the future, these limitations may be overcome, leading to their safe and effective use in humans, which will help put gene editing back on the right track. I am hopeful that our next generation of AAV6 vectors34, 35 and nuclease-free approach will also prove to be safe and effective for in vivo gene editing in humans.
References:
- Hossain MA, Shen Y, Knudson I, Thakur S, Stees JR, Qiu Y, Pave BS, Peterson KR, Bungert J. Activation of fetal g-globin gene expression via direct protein delivery of synthetic zinc-finger DNA-binding domains. Mol Ther Nucl Acids, 5: e378, 2016.
- Li B, Zhu X, Hossain MA, Guy CR, Xu H, Bungert J, Pace BS. Fetal hemoglobin induction in sickle erythroid progenitors using a synthetic zinc finger DNA-binding domain. Haematologica, 103: e384-387, 2018.
- Song L, Kauss MA, Kopin E, Chandra M, Ul-Hassan T, Miller E, Jayandharan GR, Rivers AE, Aslanidi GV, Ling C, Li, B, Ma W, Li X, Andino LM, Zhong L, Tarantal AF, Yoder MC, Wong Jr. KK, Tan M, Chatterjee S, Srivastava A. Optimizing the transduction efficiency of capsid-modified AAV6 serotype vectors in primary human hematopoieitic stem cells in vitro and in a xenograft mouse model in vivo. Cytotherapy, 15: 986-996, 2013.
- Srivastava CH, Samulski RJ, Lu L, Larsen SH, Srivastava A. Construction of a recombinant human parvovirus B19: Adeno-associated virus 2 DNA inverted terminal repeats are functional in an AAV-B19 hybrid virus. Proc Natl Acad Sci, USA, 86: 8078-8082, 1989.
- Wang X-S, Yoder MC, Zhou SZ, Srivastava A. Parvovirus B19 promoter at map unit 6 confers autonomous replication competence and erythroid specificity to adeno-associated virus 2 in primary human hematopoietic progenitor cells. Proc Natl Acad Sci, USA, 92: 12416-12420, 1995.
- Ponnazhagan S, Wang X-S, Woody MJ, Luo F, Kang LY, Nallari ML, Munshi NC, Zhou SZ, Srivastava A. Differential expression from p6 promoter of parvovirus B19 in human cells following plasmid transfection and recombinant adeno-associated virus 2 (AAV) infection: Human megakaryocytic leukaemia cells are non-permissive for AAV infection. J Gen Virol, 77: 1111-1122, 1996.
- Zhou SZ, Li Q, Stamatoyannopoulos G, Srivastava A. Adeno-associated virus 2-mediated transduction and erythroid cell-specific expression of a human -globin gene. Gene Ther, 3: 223-229, 1996.
- Kurpad C, Mukherjee P, Wang X-S, Ponnazhagan S, Li L, Yoder MC, Srivastava A. Adeno-associated virus 2-mediated transduction and erythroid lineage-restricted expression from parvovirus B19p6 promoter in primary human hematopoietic progenitor cells. Journal of Hematotherapy & Stem Cell Research, 8: 585-592, 1999.
- Song L, Li X, Jayandharan GR, Wang Y, Aslanidi GV, Ling C, Zhong L, Gao G, Yoder MC, Ling C, Tan M, Srivastava A. High-efficiency transduction of primary human hematopoietic stem cells and erythroid lineage-restricted expression by optimized AAV6 serotype vectors in vitro and in a murine xenograft model in vivo. PLoS One, 8: e58757. 2013.
- Frangoul H, Altshuler D, Cappellini MD, Chen Y-S, Domm J, Eustace BK, Foell J, de la Fuente J, Grupp S, Handgretinger R, Ho TW, Kattamis A, Kernytsky A, Lekstrom-Himes J, Li AM, Locatelli F, Mapara MY, de Montalembert M, Rondelli D, Sharma A, Sheth S, Soni S, Steinberg MH, Wall D, Yen A, Corbacioglu S. CRISPR-Cas9 Gene Editing for Sickle Cell Disease and β-Thalassemia. N Engl J Med, 384: 252-260, 2021.
- Samuelson C, Radtke S, Zhu H, Llewellyn M, Fields E, Cook S, Huang M-L W, Jerome KR, Kiem H-P, Humbert O. Multiplex CRISPR/Cas9 genome editing in hematopoietic stem cells for fetal hemoglobin reinduction generates chromosomal translocations. Mol Ther Clin Dev, 23: 507-523, 2021.
- Tsuchida CA, Brandes N, Bueno R, Trinidad M, Mazumder T, Yu B, Hwang B, Chang C, Liu J, Sun Y, Hopkins CR, Parker KR, Qi Y, Satpathy A, Stadtmauer EA, Cate JHD, Eyquem J, Fraietta JA, June CH, Chang HY, Ye CJ, Doudna JA. Mitigation of chromosome loss in clinical CRISPR-Cas9-engineered T cells. Cell 186: 4567-4582, 2023.
- Newby GA, Yen JS, Woodard KJ, Mayuranatham T, Lazzorotto CR, Li Y, Sheppard-Tillman H, Porter SN, Yao Y, Mayberry K, Everette KA, Jang Y, Podracky CJ, Thaman E, Lechauve C, Sharma A, Henderson JM, Richter MF, Zhao KT, Miller AM, Wang T, Koblan LW, McCaffrey AP, Tisdale JF, Kalfa TA, Pruett-Miller SM, Tsai SQ, Weiss MJ, Liu DR. Base editing of haematopoietic stem cells rescues sickle cell disease in mice. Nature, 595: 295-302, 2021.
- Everette KA, Newby GA, Levine RM, Mayberry K, Jang Y, Mauranatham T, Nimmagadda N, Dempsey E, Li Y, Bhoopalan SV, Liu X, Davis JR, Nelson AT, Chen PJ, Sousa AA, Cheng Y, Tisdale JF, Weiss MJ, Yen JS, Liu DR. Ex vivo prime editing of patient haematopoietic stem cells rescues sickle-cell disease phenotypes after engraftment in mice. Nature Biomed Engin, 7: 616-628, 2023.
- Fiumara M, Ferrari S, Omer-Javed A, Beretta S, Albano L, Canarutto D, Varesi A, Gaddoni C, Brombin C, Cugnata F, Zonari E, Naldini MM, Barcella M, Gentner B, Merelli I, Naldini L. Genotoxic effects of base and prime editing in human hematopoietic stem cells. Nature Biotechnol., 42: 877-891, 2024.
- Duan D, Sharma P, Yang J, Yue Y, Dudus L, Zhang Y, Fisher KJ, Engelhardt JF. Circular intermediates of recombinant adeno-associated virus have defined structural characteristics responsible for long-term episomal persistence in muscle tissue. J Virol, 72: 8568-8577, 1998.
- Nakai H, Fuess S, Storm TA, Meuse LA, Kay MA. Free DNA ends are essential for concatemerization of synthetic double-stranded adeno-associated virus vector genomes transfected into mouse hepatocytes in vivo. Mol Ther, 7: 112-121, 2003.
- Song S, Lu S, Choi Y-K, Han Y, Tang Q, Zhao Z, Berns KI, Flotte TR. DNA-dependent PK inhibits adeno-associated virus DNA integration. Proc Natl Acad Sci, USA, 101: 2112-2116, 2004.
- Miao CH, Snyder RO, Schowalter DB, Patijn GA, Donahue B, Winther B, Kay MA. The kinetics of rAAV integration in the liver. Nat Genet, 19: 13-15, 1998.
- Clark KR, Sferra TJ, Lo W, Qu G, Chen R, Johnson PR. Gene transfer into the CNS using recombinant adeno-associated virus: analysis of vector DNA forms resulting in sustained expression. J Drug Target, 7: 269-283, 1999.
- Nakai H, Montini E, Fuess S, Storm TA, Grompe M, Kay MA. AAV serotype 2 vectors preferentially integrate into active genes in mice. Nat Genet, 34: 297-302, 2003.
- Zhong L, Li W, Li Y, Zhao W, Wu J, Li B, Maina N, Bischof D, Qing K, Weigel-Kelley KA, Zolotukhin, Warrington Jr KH, Li X, Slayton WB, Yoder MC, Srivastava A. Evaluation of primitive murine hematopoietic stem and progenitor cell transduction in vitro and in vivo by recombinant adeno-associated virus vector serotypes 1 through 5. Hum Gene Ther, 17: 321-333, 2006.
- Zhong L, Zhao W, Wu J, Maina N, Han Z, Srivastava A. Adeno-associated virus-mediated gene transfer in hematopoietic stem/progenitor cells as a therapeutic tool. Curr Gene Ther, 6: 683-698, 2006.
- Maina N, Han Z, Li X, Hu Z, Zhong L, Bischof D, Weigel-Van Aken KA, Slayton WB, Yoder MC, Srivastava A. Recombinant self-complementary adeno-associated virus serotype vector-mediated hematopoietic stem cell transduction and lineage-restricted, long-term transgene expression in a murine serial bone marrow transplantation model. Hum Gene Ther, 19: 376-383, 2008.
- Han Z, Zhong L, Maina N, Hu Z, Li L, Chouthai NS, Bischof D, Weigel-Van Aken KA, Slayton WB, Yoder MC, Srivastava A. Stable integration of recombinant adeno-associated virus vector genomes after transduction of murine hematopoietic stem cells. Hum Gene Ther, 19: 267-278, 2008.
- Nguyen GN, Everett JK, Kafle S, Roche AM, Raymond HE, Leiby J, Wood C, Assenmacher C-A, Merricks EP, Long CT, Kazazian HH, Nichols TC, Bushman FD, Sabatino, DE. A long-term study of AAV gene therapy in dogs with hemophilia A identifies clonal expansions of transduced liver cells. Nat Biotechnol, 39: 47-55, 2021.
- Greig JA, Martin KM, Brenton C, Lamontagne RJ, Zhu Y, He Z, White J, Zhu J-X, Chichester JA, Zheng Q, Zhang Z, Bell P, Wang L, Wilson JM. Integrated vector genomes may contribute to long-term expression in primate liver after AAV administration. Nat Biotechnol, 42: 1232-1242, 2024.
- Reiss UM, Davidoff AM, Tuddenham EGD, Chowdary P, McIntosh J, Muczynski V, Journou M, Simini G, Ireland L, Mohamed S, Riddell A, Pie AJ, Hall A, Quaglia A, Mangles S, Mahalangu J, Haley K, Recht M, Shen Y-M, Halka KG, Fortner G, Morton CL, Gu Z, Hayden RT, Neufeld EJ, Okhomina VI, Kang G, Nathwani AC. Sustained Clinical Benefit of AAV Gene Therapy in Severe Hemophilia B. N Engl J Med, 392: 2226-2234, 2025.
- Frangoul H, Locatelli F, Sharma A, Bhatia M, Mapara, M, Molinari L, Wall D, Liem R, Telfer P, Shah AJ, Cavazzana M, Corbacioglu S, Rondelli M, Meisel R, Dedeken L, Lobitz S, de Montalembert M, Steinberg MH, Walters MC, Eckrich MJ, Imren S, Bower L, Simard C, Zhou W, Xuan F, Morrow PK, Hobbs WE, Grupp SA. Exagamglogene Autotemcel for Severe Sickle Cell Disease. N Engl J Med, 390: 1649-1662, 2024.
- Locatelli F, Lang P, Wall D, Meisel R, Corbacioglu S, Li AM, de la Fuente J, Shah AJ, Carpenter B, Kwiatkowski JL, Mapara M, Liem, Cappellini MD, Algeri M, Kattamis A, Sheth S, Grupp S, Handgretinger R, Kohli P, Shi D, Ross L, Bobruff Y, Simard C, Zhang L, Morrow PK, Hobbs WE, Frangoul H. Exagamglogene Autotemcel for Transfusion-Dependent β-Thalassemia. N Engl J Med, 390: 1663-1676, 2024.
- Cohn DM, Gurugama P, Magerl M, Katelaris CH, Launay D, Bouillet L, Petersen RS, Lindsay K, Aygören-Pürsün E, Maag D, Butler JS, Shah MY, Golden A, Xu Y, Abdelhady AM, Lebwohl D, Longhurst HJ. CRISPR-Based Therapy for Hereditary Angioedema. N Engl J Med, 392: 458-467, 2025.
- Musunuru K, Grandinette SA, Wang X, Hudson TR, Briseno K, Berry AM, Hacker JL, Hsu A, Silverstein RA, Hille LT, Ogul AN, Robinson-Garvin NA, Small JC, McCague S, Burke SM, Wright CM, Bick S, Indurthi V, Sharma S, Jepperson M, Vakulskas CA, Collingwood M, Keogh K, Jacobi A, Sturgeon M, Brommel C, Schmaljohn E, Kurgan G, Osborne T, Zhang H, Kinney K, Rettig G, Barbosa CJ, Semple SC, Tam YK, Lutz C, George LA, Kleinstiver BP, Liu DR, Ng K, Kassim SH, Giannikopoulos P, Alameh M-G, Urnov FD, Ahrens-Nicklas RC. Patient-Specific In Vivo Gene Editing to Treat a Rare Genetic Disease. N Engl J Med 392: 2235-2243, 2025.
- Roberts R. Alternatives to CRISPR-Cas9: Nucleases for next-gen therapy. Cell & Gene, 1-4, 2025.
- Ling C, Bhukhai K, Yin Z, Tan M, Yoder MC, Leboulch P, Payen E, Srivastava A. High-efficiency transduction of primary human CD34+ hematopoietic stem/progenitor cells by AAV6 serotype vectors: Strategies for overcoming donor-variation and implications in genome editing. Scientific Reports, 6, 35495, 2016.
- Yang H, Qing K, Keeler GD, Yin L, Mietzsch M, Ling C, Hoffman BE, Agbandje-McKenna M, Tan M, Wang W, Srivastava A. Enhanced transduction of human hematopoietic stem cells by AAV6 vectors: Implications in gene therapy and genome editing. Molecular Therapy-Nucleic Acids, 20: 451-458, 2020.
About The Expert:
 Arun Srivastava, Ph.D., is George H. Kitzman Professor of Genetics and Chief of Division of Cellular and Molecular Therapy at the University of Florida College of Medicine in the pediatrics, molecular genetics & microbiology departments and the college’s Powell Gene Therapy Center. He was a founder of the first AAV gene therapy company, Avigen, in 1992, and later cofounded Lacerta Therapeutics, and more recently, sAAVient Therapeutics, and nAAVigen Therapeutics. He serves on an NIH study section and the editorial boards of several academic journals. He has published more than 224 peer-reviewed articles, book chapters, reviews, and other articles. He received his Ph.D. from the Indian Institute of Science in Bangalore and completed postdoctoral training at Memorial Sloan-Kettering Cancer Center. He was recently elected as a Fellow of the American Society for the Advancement of Science.
Arun Srivastava, Ph.D., is George H. Kitzman Professor of Genetics and Chief of Division of Cellular and Molecular Therapy at the University of Florida College of Medicine in the pediatrics, molecular genetics & microbiology departments and the college’s Powell Gene Therapy Center. He was a founder of the first AAV gene therapy company, Avigen, in 1992, and later cofounded Lacerta Therapeutics, and more recently, sAAVient Therapeutics, and nAAVigen Therapeutics. He serves on an NIH study section and the editorial boards of several academic journals. He has published more than 224 peer-reviewed articles, book chapters, reviews, and other articles. He received his Ph.D. from the Indian Institute of Science in Bangalore and completed postdoctoral training at Memorial Sloan-Kettering Cancer Center. He was recently elected as a Fellow of the American Society for the Advancement of Science.