
FY2017 FDA Drug Inspection Observations And Trends
By Barbara Unger, Unger Consulting Inc.


A comprehensive GMP intelligence program includes monitoring of health authority enforcement actions, including FDA Form 483s, Establishment Inspection Reports, warning letters, recalls, import alerts, consent decree agreements, and EU reports of GMDP noncompliance. This article presents the most recent publication of GMP drug product inspection data from CDER, which addresses drug inspections conducted in FY2017. We look at five years of data for the FDA, examine data from 2017, and evaluate five years’ worth of trends in GMP inspection enforcement. The CDER drug inspection observations supplement the information previously published describing CDER Drug GMP warning letters issued in FY2017.
The presentation of data herein differs from data presented on the FDA website, even though it uses the same raw data. For example, I combine all observation listings that cite 21 CFR 211.192 into a single value. In contrast, the FDA data presents five line items for §211.192, and each identifies the number of times FDA cited the regulation. Consolidating the citations leads to different conclusions. In the FDA listing, the most frequently cited item is §211.22(d), “procedures not in writing, fully followed.” Considering the total number of times that the FDA cited §211.192, §211.188, and §211.160(b), they become numbers one, two, and three, respectively, with §211.22(d) becoming the fourth most frequent citation. The FDA uses the term “frequency,” which appears to be the number of times the agency identified a specific citation in its tabulation.
The FDA’s data includes only Form 483s issued through its electronic system; it does not include Form 483s issued to API manufacturers, because §211 cannot be applied to those manufacturers by law, or Form 483s issued outside of the electronic system. Thus, the data does not represent the FDA's complete collection of inspection observations for the year.
Executive Summary
- The number of Form 483s included in this analysis remained reasonably consistent over the past five fiscal years, ranging between 645 and 694 inspections. (Table 1, row 2)
- The number of observations citing failures in investigation of discrepancies (§211.192) remains at the top of this list over the past five years, though it tied for first place last year with §211.42(c). We as an industry cannot seem to get this investigation activity quite right. (Table 1)
- In general, the regulations cited, and their relative order, have remained reasonably constant over the past fiscal years. Five areas, however, saw a marked change, two with increases and three with decreases, between FY2016 and FY2017. (Table 2 and Figure 2)
FDA Form 483 Inspection Observations
Table 1 shows only the 15 most frequent inspection observation citations, while the tabulation on the FDA website shows all citations used in the fiscal year. Table 1 presents those observations from the highest to lowest number for 2017, modified as described in the Introduction section of this paper. Both Table 1 and Figure 1 show consistency in the years between FY2013 and 2017 with respect to the overall identity of inspection observations.
Investigations, covered in §211.192, remain at the top of the list for all five years, albeit tied last year for that position. Figure 1 shows a graphic representation of the data from Table 1. In five instances, highlighted in gray in Table 1, the number of times the FDA cited a specific regulation changed markedly in FY2017.
Table 2 shows only the five marked citation differences from FY2016 and FY2017. Three regulations decreased in the number of times they were cited in FY2017 compared with 2016: §211.42(c) (facilities shall include defined areas of sufficient size), §211.166(a) (stability testing), and §211.67(a) (equipment cleaning, sanitization, and sterilization). These three regulations continued a downward trend that started in FY2015, as shown in both Table 2 and Figure 2. Two regulations saw a significant increase in FY2017 over FY 2016: §211.188 (Master production and control records) and §211.160(b) (lab controls with scientifically sound specifications). The increase in citations of §211.188 was exceptional. The FDA cited this regulation an average of 100 times in the four previous years, with values ranging between 74 and 114; in 2017 it cited this regulation 208 times. For §211.160(b), the value increased markedly, from 133 in 2016 to 207 in 2017. The values between 2013 through 2016, however, were more highly variable and ranged from a low of 133 in 2016 to a high of 246 in 2015.
I cannot identify the reason(s) for the differences in the number of times the FDA identified the five citations. The FDA increasingly focused on over-the-counter (OTC) drug manufacturers in 2017, and many of these inspections cite either lack of, or incomplete, batch records. Emphasis on laboratory controls and specifications may have increased for similar reasons. I’ve not seen hard data to suggest reasons why any sections within §211 have either decreased or increased in number.
Table 1: Drug GMP Inspections, §211 Citations
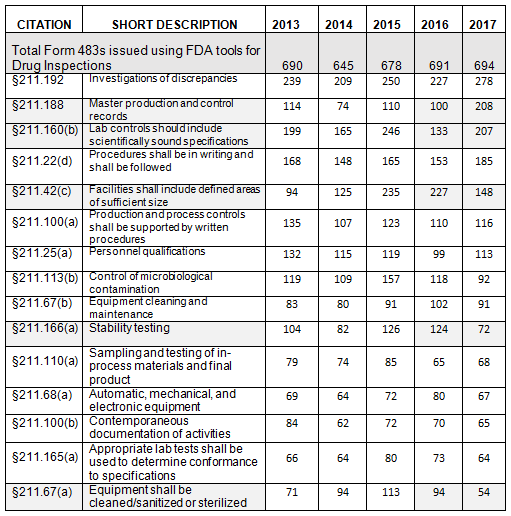
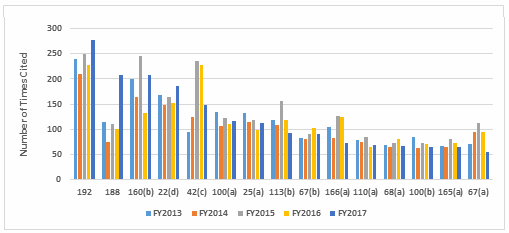
Figure 1: Top 15 citations
Table 2: FDA Drug Regulation Citations in FY2016 and FY 2017 Inspections for Selected Areas
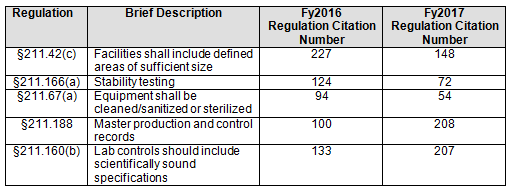
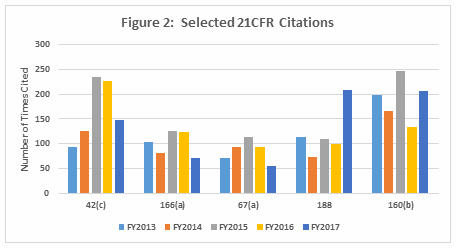
Figure 2: Selected 21CFR citations
Conclusions
For those who use inspection observations to monitor and improve their quality systems, the FDA’s annual data provides ample resources against which firms can measure their potential vulnerabilities and gauge the probable focus areas during upcoming GMP inspections. The FDA’s focus on investigations consistently scored first on the list of inspection observations over the past five years. The industry seems to fail here for several reasons:
- Firms fail to identify deviations and the need for investigations,
- Firms fail to include assessment of all lots that may be impacted in the investigation to identify root cause,
- Firms fail to identify a root cause, and
- Firms lack science-based conclusions and follow-up, including evaluation of the effectiveness of any corrective actions.
The dramatic increase in the number of times that the FDA cited §211.188 in 2017 should cause smaller firms and OTC manufacturers to ensure they establish adequate controls for batch record development and execution.
And finally, the lack of adequate written procedures and responsibilities for the quality unit, §211.22(d), remains a very consistent citation over the five years addressed herein. Form 483 observations that include text such as “The Quality Unit is Inadequate…” often result in additional enforcement actions including warning letters.
About The Author:
 Barbara Unger formed Unger Consulting, Inc. in December 2014 to provide GMP auditing and regulatory intelligence services to the pharmaceutical industry, including auditing and remediation in the area of data management and data integrity. Her auditing experience includes leadership of the Amgen corporate GMP audit group for APIs and quality systems. She also developed, implemented, and maintained the GMP regulatory intelligence program for eight years at Amgen. This included surveillance, analysis, and communication of GMP related legislation, regulations, guidance, and industry compliance enforcement trends. Barbara was the first chairperson of the Rx-360 Monitoring and Reporting work group (2009 to 2014) that summarized and published relevant GMP and supply chain related laws, regulations, and guidance. She also served as the chairperson of the Midwest Discussion Group GMP-Intelligence sub-group from 2010 to 2014. Barbara is currently the co-lead of the Rx-360 Data Integrity Working Group.
Barbara Unger formed Unger Consulting, Inc. in December 2014 to provide GMP auditing and regulatory intelligence services to the pharmaceutical industry, including auditing and remediation in the area of data management and data integrity. Her auditing experience includes leadership of the Amgen corporate GMP audit group for APIs and quality systems. She also developed, implemented, and maintained the GMP regulatory intelligence program for eight years at Amgen. This included surveillance, analysis, and communication of GMP related legislation, regulations, guidance, and industry compliance enforcement trends. Barbara was the first chairperson of the Rx-360 Monitoring and Reporting work group (2009 to 2014) that summarized and published relevant GMP and supply chain related laws, regulations, and guidance. She also served as the chairperson of the Midwest Discussion Group GMP-Intelligence sub-group from 2010 to 2014. Barbara is currently the co-lead of the Rx-360 Data Integrity Working Group.
Barbara received a bachelor's degree in chemistry from the University of Illinois at Urbana-Champaign. You can contact her at bwunger123@gmail.com.