
From QM To QMM For AI-Assisted Pharmaceutical Manufacturing Operations
By Gautam Shah, president, U.S. operations, Aavis Pharmaceuticals, a subsidiary of Senores; and Ajay Babu Pazhayattil, Ph.D., president, cGMPWorld

The FDA’s focus has shifted from simply tracking quality metrics1,2,3,4,5 to promoting quality management maturity (QMM), the idea that a strong quality culture matters more than just having the right forms in place. Recent pilot programs, including the 2020–2022 QMM pilots and the 2024 Prototype Assessment Protocol Evaluation Program,6, 7 reflect this change by looking at how quality is actually woven within daily management decisions, knowledge sharing, and operations. A big part of this is building a real quality culture where employees are comfortable bringing up concerns, management treats mistakes as chances to learn rather than blame, and decisions always put patient safety and product quality first. Still, making that happen isn’t easy. It means surmounting resistance from leadership, breaking down silos between departments, and moving past a box-checking view of compliance. To help, the FDA is encouraging technology that supports data integrity and rewarding manufacturers that show reliable, high-quality operations. Making that shift, though, means changing how an organization operates, starting from how teams communicate to how quality decisions are made and who gets to make them. The impact of machine learning (ML) and artificial intelligence (AI) on pharmaceutical quality systems is becoming increasingly significant. Their application in areas such as automated visual inspection (AVI) directly impacts product quality decisions, which in turn demands novel approaches to quality management. The expanding use of generative AI introduces additional complexity, particularly around data integrity and regulatory compliance.
Real-Time Monitoring, A Foundation For Advanced Analytics And AI
Continued process verification (CPV, Stage 3) provides a structured, lifecycle framework for real-time and near real-time monitoring of process parameters and quality attributes. The data-rich environment can be leveraged for advanced analytics and AI applications, enabling early detection of variability, deeper process understanding, predictive and robust control strategies that directly support continuous improvement. ICH (Q8, Q11, Q12, Q14) emphasizes lifecycle knowledge management and ongoing monitoring for science- and risk-based decision making, aligning with the adoption of digital and analytical technologies..
Solutions Addressing Emerging Challenges: A Maturity Indicator
Traditional documentation systems have long been vulnerable to the widespread use of copy-paste, leading to the propagation of errors such as outdated or irrelevant information that can result in regulatory citations. Alarmingly, FDA warning letters have cited data integrity discrepancies, including those stemming from copy-paste errors.9
The potential for personnel to use widely available generative AI tools amplifies existing data integrity challenges. AI introduces novel risks that require proactive quality assurance strategies. AI can overwrite metadata and obscure authorship and timestamps, potentially compromising traceability. Generative AI can propagate false information by automating flawed data and templates. It can also enable the widespread reuse of error-prone data without supporting evidence and embed it in records. All of these violate basic ALCOA+ principles.
Additionally, AI introduces its own set of QA challenges, such as generating hallucinated compliance data that appears valid but lacks a factual basis. Autonomous updates generated by AI models could result in undocumented changes that escape traditional audit trails, thereby violating regulations such as 21 CFR Part 11.
The reach and scale of generative AI in pharma can lead to the rapid spread of misinformation it produces across the organization's quality system documents. To address this risk, a robust centralized QA framework with AI safeguards, along with traditional compliance controls, is required. It is essential to verify the accuracy of the content, especially in technical or regulated domains, where AI may confidently present incorrect, outdated, or misleading information. Today, QA needs to carefully examine whether data, references, and conclusions are supported by real data and sources and ensure the report aligns with industry standards and regulatory expectations. They need to watch for signs of speculation, such as overly confident claims without evidence/references. Involving a subject matter expert and a forensic IT solution in a review may now be essential to catch nuanced errors that generative AI tools may introduce when personnel use them intentionally or unintentionally.
For high-risk documents such as stability reports or regulatory submissions, stricter measures are needed, including mandatory AI-generation detection and sign-off. During the review phase, an automated or manual prescreening step that cross-checks data against source systems and that flags discrepancies is best. This is followed by a tiered human review involving specialists. Detection of AI-generated content should be a standard step, as it is crucial for maintaining integrity and compliance.
There are specialized tools/models that are commercially available to detect AI-assisted writing at various levels of complexity. Organizations' QA teams must stay abreast of the latest tools, given that AI models evolve and some tools can become less reliable over time. Best practices include combining automated checks with human review, training personnel on the ethical use of AI, and leveraging tools for cross-verification. Once QA detects and flags documents and data generated by AI, notifications to departments, actions, and maintaining metrics are essential to demonstrate maturity. By integrating control measures, QA can balance innovation and accelerate document preparation processes while rigorously reducing error rates.
While regulatory intelligence helps organizations stay ahead of compliance risks by leveraging insights from other organizations, QMM empowers them to rapidly detect internal gaps and respond with agility. QMM represents a transformative element of pharmaceutical quality management, integrating quality metrics, quality culture, monitoring, and AI governance into a unified data-driven framework. By consolidating real-time dashboards, predictive analytics/monitoring solutions, and AI checks, we enable proactive QA decision-making and preempt regulatory compliance risks, strategic for any organization. The one major enabler is knowledge management. QMM doesn’t just ensure compliance; it preempts and thus redefines quality as a competitive business advantage, ensuring patient safety, supporting innovation, maintaining regulatory trust, and delivering measurable outcomes. The ROI is reaped as the system matures, simultaneously reducing risks and accelerating operational excellence.
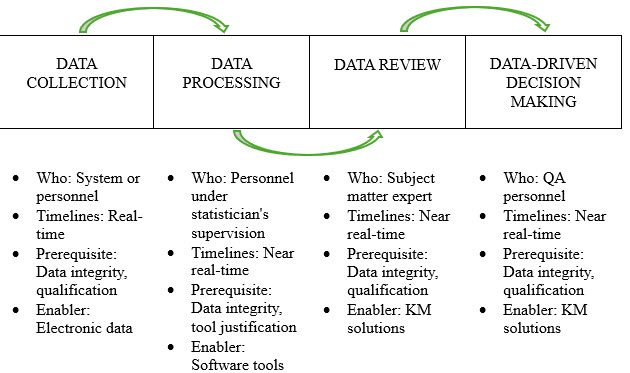
Figure 1: Mapping The Cycle for Rapid and Robust QA Decision Making
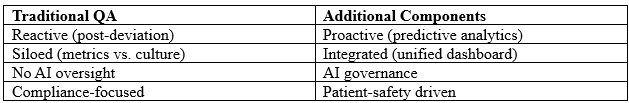
Table 1: Outperforming Traditional QA
The evolving landscape of pharmaceutical quality assurance, driven by frameworks, demands a harmonized and future-ready QMM framework. Organizations often approach metrics, culture, and digital controls as separate initiatives. Regular meetings between cross-functional quality councils can ensure alignment. Organizations must adopt a proactive, harmonized approach (Table 1) to ensure sustainable quality excellence while mitigating risks to patient safety and regulatory compliance. Organizations can no longer afford siloed approaches to quality management. It is essential to establish a core team to oversee metrics, culture, signals, and AI governance and to support quick-turnaround, data-driven decision-making from QA. An enabled quality unit allows for organizational alignment from the shop floor to the C-suite (Figure 1). The time for reactive quality is over; the era of patient-centric predictive quality excellence, QMM, has begun.
References
- https://www.federalregister.gov/documents/2015/07/28/2015-18448/request-for-quality-metrics-notice-of-draft-guidance-availability-and-public-meeting-request-for
- https://www.federalregister.gov/documents/2016/11/25/2016-28332/submission-of-quality-metrics-data-draft-guidance-for-industry-availability-request-for-comments
- https://item.unisg.ch/divisions/production-management/focus/operational-excellence/quality-metrics-research/
- https://www.fda.gov/drugs/pharmaceutical-quality-resources/quality-metrics-drug-manufacturing#
- https://www.fda.gov/media/131130/download?attachment
- https://www.fda.gov/media/171705/download?attachment
- https://www.federalregister.gov/documents/2024/01/25/2024-01423/voluntary-quality-management-maturity-prototype-assessment-protocol-evaluation-program#
- www.fda.gov/files/drugs/published/Process-Validation--General-Principles-and-Practices.pdf
- https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/compliance-actions-and-activities/warning-letters
About The Authors:
 Gautam Shah brings over 30 years of experience in the global pharmaceutical industry, including 20 years in the United States and 10 years in India. He has held senior leadership roles at leading pharmaceutical companies such as Cipla, Sun Pharma, Caraco Pharmaceutical Labs, Dow Corning Corp, Nesher Pharmaceuticals, Med-Pharmex, and Accord Healthcare. He holds a bachelor’s degree in pharmacy, a post-graduate diploma in business administration, and is a certified Six Sigma Black Belt. His expertise spans quality assurance, compliance, manufacturing, operations, and strategic leadership. At Senores, he is the president of U.S. operations at Aavis Pharmaceuticals, a subsidiary of Senores Pharmaceuticals Limited and leads the overall U.S. strategy, operations, and growth initiatives.
Gautam Shah brings over 30 years of experience in the global pharmaceutical industry, including 20 years in the United States and 10 years in India. He has held senior leadership roles at leading pharmaceutical companies such as Cipla, Sun Pharma, Caraco Pharmaceutical Labs, Dow Corning Corp, Nesher Pharmaceuticals, Med-Pharmex, and Accord Healthcare. He holds a bachelor’s degree in pharmacy, a post-graduate diploma in business administration, and is a certified Six Sigma Black Belt. His expertise spans quality assurance, compliance, manufacturing, operations, and strategic leadership. At Senores, he is the president of U.S. operations at Aavis Pharmaceuticals, a subsidiary of Senores Pharmaceuticals Limited and leads the overall U.S. strategy, operations, and growth initiatives.
 Ajay Babu Pazhayattil, Ph.D., is an accomplished management consultant and industrial pharmacist with extensive experience spanning solid oral, sterile, and API sectors. He is the founder of cGMP World. Pazhayattil has held key leadership positions with prominent North American brands, generic manufacturers, and CDMOs. His roles include vice president of scientific and regulatory affairs at Capcium, quality director at Eurofins, and associate director at Apotex. He plays a pivotal role in guiding organizations through remediation efforts related to U.S. FDA 483 observations and warning letters. He has been a lead author and contributor to guidance documents published by industry organizations such as AAPS, PDA, ISPE, and RAPS.
Ajay Babu Pazhayattil, Ph.D., is an accomplished management consultant and industrial pharmacist with extensive experience spanning solid oral, sterile, and API sectors. He is the founder of cGMP World. Pazhayattil has held key leadership positions with prominent North American brands, generic manufacturers, and CDMOs. His roles include vice president of scientific and regulatory affairs at Capcium, quality director at Eurofins, and associate director at Apotex. He plays a pivotal role in guiding organizations through remediation efforts related to U.S. FDA 483 observations and warning letters. He has been a lead author and contributor to guidance documents published by industry organizations such as AAPS, PDA, ISPE, and RAPS.