
4 Focus Areas To Expand The Success Of Antibody-Drug Conjugates
By Stuart D. Barnscher, director, preclinical programs, ADC therapeutic development, Zymeworks Inc.

More than a century ago, Nobel laureate Paul Ehrlich, widely recognized as the founder of chemotherapy, first theorized about the development of “magic bullets” able to precisely target diseased tissues while sparing normal healthy tissues. Building on this concept, in recent years researchers around the world have focused their efforts on antibody-drug conjugates (ADCs) as potential magic bullets for the treatment of many types of cancer. After decades of technological advancements and learnings to better understand these complex molecules, ADCs have now entered their golden age.
ADCs represent a significant advance in therapeutic engineering. They combine an antibody, a linker, and a payload designed into a single molecule for delivery to a specific target. (Figure 1 below.)
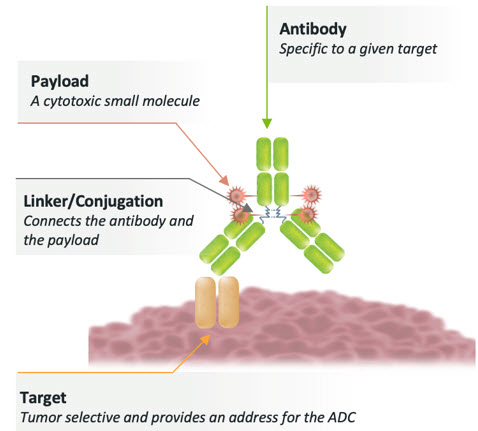
Figure 1: Architecture of an ADC
Built on the pioneering work that led to the successful development of the first approved ADCs (Adcedtris, Kadcyla, Mylotarg, and Besponsa), a total of eight ADCs were approved by the FDA between 2019 and 2022 and there are additional promising ADCs in late-stage clinical development. (Figure 2 below.)
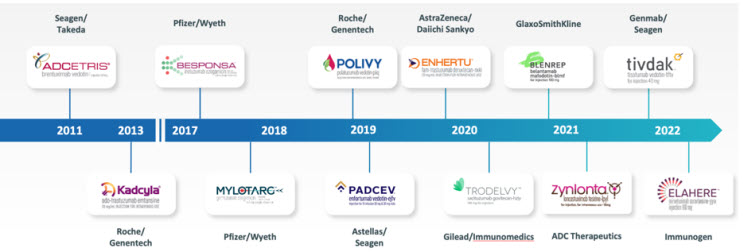
Figure 2: FDA approval timeline for antibody-drug conjugates
The 12 total approved ADCs represent advances in the treatment of both liquid and solid tumors using a variety of payload mechanisms with a range of potencies. The more potent DNA-damaging agents, like calicheamicin and pyrrolobenzodiazepine (PBD) dimers, have been successful in liquid tumors targeting CD22, CD33, and CD19, while the slightly less potent microtubule inhibitors have seen success in liquid tumors targeting CD30, BCMA, and CD79b and in solid tumors targeting HER2, tissue factor (TF), Nectin-4, and folate receptor alpha (FR⍺). The relatively lower potency topoisomerase 1 inhibitors have been successful in solid tumors targeting HER2 and TROP2. (Figure 3 below.)

Figure 3: Approved ADCs categorized by liquid or solid tumor settings and payload mechanism
Despite these recent successes, discontinuation rates for ADCs remain high across multiple payload mechanisms, especially for the more established payload classes like the auristatins, maytansinoids, and PBD-dimers. The more nascent camptothecin class of payload is on a promising trajectory with two approved ADCs, Enhertu and Trodelvy, and very few discontinued programs to date. (Figure 4 below.)
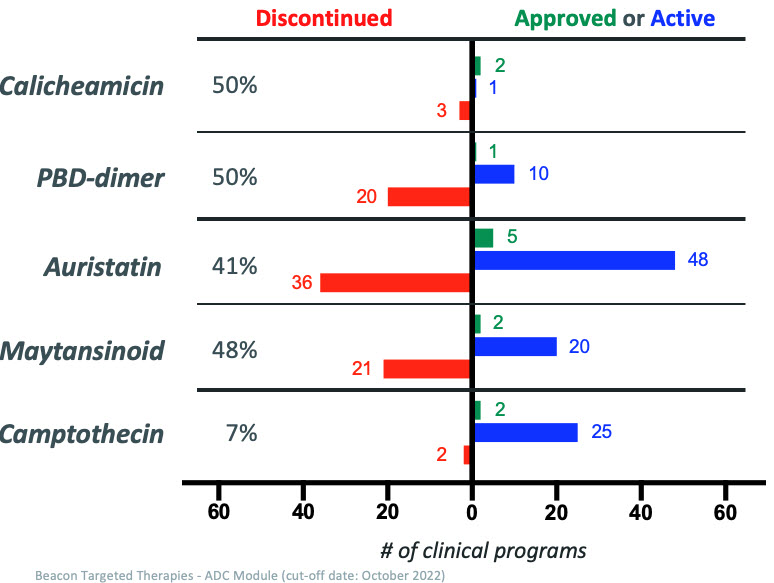
Figure 4: Approved, active, and discontinued clinical ADC programs categorized by payload class
While ADC programs may fail for different reasons, in most cases the factors involved ultimately result in either on-target toxicity due to target expression on normal tissues or lack of efficacy despite achieving the maximum tolerated dose (MTD) as dictated by the payload (often termed “platform MTD”). Very few ADC programs are discontinued due to on-target toxicity and most program discontinuations occur due to a lack of efficacy at the platform MTD. A framework for increasing the likelihood that an ADC program will be successful is based on selection and combination of the right design criteria for each ADC component such that meaningful patient benefit can be achieved at the platform MTD. The target, the antibody, the linker, the conjugation method, and the payload all need to be considered and optimized. In Table 1 and the discussion below, I distinguish between our industry’s conventional strategies and the novel strategies we can employ to design better ADCs.
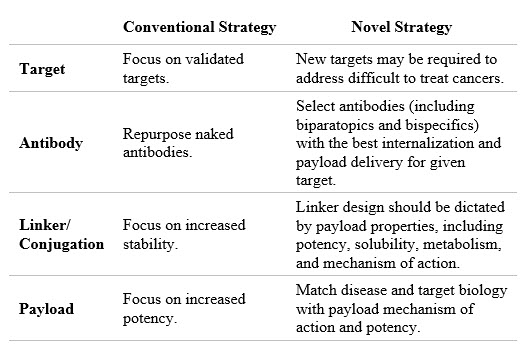
Table 1: Strategy to reduce discontinuation levels
The Target
When considering the target, researchers need to focus on both the target expression profile and the target biology, which will dictate how to select the antibody binding properties and the payload potency and mechanism of action. Tumor-specific targets are rare, therefore understanding the prevalence and distribution of the target in normal tissues is important. High normal tissue expression may dictate that a lower potency payload is required. Similarly, the highest affinity antibody may not be the best candidate if there are normal tissue expression concerns for the target. Known targets are highly competitive. For popular targets like HER2 and FR⍺ there are dozens of ADCs at various stages of preclinical and clinical development. These types of targets may still be actionable when armed with the right technologies and the right design; however, as researchers continue to address difficult to treat cancers, we will likely need to explore targets with less validation.
The Antibody
In ADC development, researchers often fail to focus adequately on the antibody, in many cases using repurposed antibodies from failed naked antibody programs. This strategy of taking a failed naked antibody and repurposing it as a component of an ADC may not yield optimal results. While the alternative approach requires more investment in time and resources, efforts to discover and develop antibodies with enhanced binding, internalization, and payload delivery properties have the potential to improve the likelihood an ADC will be successful. This approach can leverage bi-specific and bi-paratopic strategies proven to engage targets in unique ways to drive better internalization and specificity and potentially lower the target expression threshold required for anti-tumor activity.
The Linker/Conjugation
For an ADC, researchers should focus their efforts on pairing the linker and conjugation method that matches the payload potency, solubility, metabolism, and mechanism of action. Using validated peptide-cleavable linkers can often be a good place to start as more than half of approved ADCs leverage this technology. Stochastic conjugation, either through endogenous cysteine or lysine residues, is still the gold standard and is used in all 12 FDA-approved ADCs. After more than a decade of focus on development of highly stable site-specific conjugation methods, there has yet to be an approved ADC using these technologies. Researchers should be open to development of novel linkers on a case-by-case basis dictated by payload properties and other needs dictated by the target, antibody, or therapeutic indication.
The Payload
Research thus far has demonstrated that applying a widely used payload to an antibody without considering the target or tumor biology is less likely to produce a successful ADC. This is exemplified by the large number of discontinued ADC programs using auristatin and maytansinoid payloads (Figure 4). Discontinued ADC programs using these payloads are a result of the initial success of Adcetris (an auristain payload) and Kaydcyla (a maytansinoid payload) followed by pervasive use of these payloads in numerous ADC programs without much thought given to target biology, tumor biology, or antibody properties. Researchers at Zymeworks are focusing on a novel topoisomerase-1 inhibitor ADC platform that is being deployed in three preclinical ADC programs (ZW191, ZW251, and ZW220). ADCs with this payload class have demonstrated meaningful clinical benefits to patients across diverse solid tumor indications.
In addition to the established and emerging ADC payload classes, researchers should consider development of novel payloads for application to the ADC modality. This approach has the potential to provide more opportunities to match disease and target biology with payload mechanism.
Putting It All Together
ADC development is complex, and ADCs exert their therapeutic and toxic effects through multiple mechanisms. Employing a one-size-fits-all solution for any one ADC component, including the target, antibody, linker, conjugation, or payload, can result in an ADC that falls short of providing a meaningful benefit to patients. To increase the likelihood of success, ADC development requires a design philosophy that addresses a broad range of needs in target, antibody, linker, conjugation, and payload assessments. Access to a toolbox of technologies is often required to allow for careful selection and tuning of each ADC design feature. With the recent explosion of ADC approvals changing the treatment paradigm for many patients, the future continues to look bright for this exciting therapeutic modality.
About The Author:
 Stuart D. Barnscher leads the discovery and development of novel drug candidates and ADC technologies with a focus on antibody discovery and engineering, in vitro biology, and in vivo pharmacology and pharmacokinetics at Zymeworks. He has led multiple research programs and made significant contributions to the design and development of ZW49. He was instrumental in the development of multiple ADC drug-linker technologies developed at Zymeworks, including the topoisomerase-1 inhibitor platform, the ZymeLink auristatin platform, the ZymeLink Hemiasterlin platform, and the toll-like receptor 7 (TLR-7) agonist platform. Barnscher obtained his undergraduate degree in biochemistry from the University of British Columbia.
Stuart D. Barnscher leads the discovery and development of novel drug candidates and ADC technologies with a focus on antibody discovery and engineering, in vitro biology, and in vivo pharmacology and pharmacokinetics at Zymeworks. He has led multiple research programs and made significant contributions to the design and development of ZW49. He was instrumental in the development of multiple ADC drug-linker technologies developed at Zymeworks, including the topoisomerase-1 inhibitor platform, the ZymeLink auristatin platform, the ZymeLink Hemiasterlin platform, and the toll-like receptor 7 (TLR-7) agonist platform. Barnscher obtained his undergraduate degree in biochemistry from the University of British Columbia.