
Establishing Best Practices For Risk-Based Environmental Monitoring Of Modern Drug Product Facilities
By Dawn Watson, Merck & Co., Inc.

EM programs are one of the most effective tools in monitoring the state of control of classified manufacturing areas.
Environmental monitoring (EM) data provide assurance that medicines made in classified manufacturing areas are safe for patients and supply is not interrupted due to contamination issues. An EM program is thus a vital part of the quality system in all modern drug production facilities and is the subject of scrutiny by regulatory agencies.
An EM program should provide verification that a particular area is capable of being maintained within the appropriate microbiological contamination limits required by the respective classification of that area. Data from an EM program provide information about aspects of contamination prevention such as facility design, heating, ventilation, and air conditioning (HVAC) performance, high efficiency particulate air (HEPA) filter system function, the cleaning and sanitization program, effectiveness of aseptic techniques used during interventions, personnel behavior, gowning practices, material and personnel flow, equipment used, and ongoing verification of the bio-decontamination cycles in isolators. An EM program is only reliable, however, if an appropriate number of sampling points are located in the right positions in concordance with risk levels using appropriate sampling methods.
Contamination risks in biopharmaceutical manufacturing vary depending on the type of room and the manufacturing processes used. A risk-based approach to the design of EM programs is expected by regulatory agencies (see references). It is standard practice in the industry to perform a risk assessment in the facility and to design an EM program to match the risk profile. It is a well understood regulatory expectation that facilities use scientifically justified rationales for sample locations and inspectors review supporting documentation. Key legislation and guidances are referenced below.
A Standard Is Required To Establish Best Practices
Designing an EM program is a relatively complex task; facilities vary considerably and there is inadequate specific guidance on how to design EM programs and no single standard risk assessment methodology exists. It is left to each facility and regulatory agency inspector to determine the risk factors for microbiologic contamination and the adequacy of monitoring.
Whilst there are many different tools available, there is no consensus on best practice for a risk assessment (RA). RA tools used to date have been originally developed for assessing process or product risks, such as failure modes and effects analysis (FMEA) and, in the food industry, hazard analysis and critical control points (HACCP). These RA tools are useful to a degree but are of limited use for risk-based EM as they do not include guidance on how to perform an RA systematically, which risk factors to consider, how to assess the risk factors for relative risk, and how to design an EM program according to risk level.
Historically, EM programs have evolved following a “more is better” approach, largely due to corrective actions to microbial excursions that have occurred during batch manufacturing and regulatory agency inspector observations, which often differ from one inspector to another. Without best practice guidance, existing EM programs can be difficult to explain or justify and may not be assessing the highest-risk locations with most probable microbial recoveries. Sustaining best practice over time is also more difficult without an accepted standard. Moreover, interventions to conduct EM in themselves may not be evaluated as part of the overall risk assessment. EM programs are typically reflective of traditional aseptic processing paradigms. When we compared EM programs amongst different companies and within companies, there was a substantial variation with respect to sample location, sampling frequency, and sampling method. In fact, without a framework and common language, it was even difficult to discuss and compare EM programs. This wide variation leads to further uncertainty about whether the EM program is fit for purpose. The rationale of an EM program should be easy to follow and compare with best practice.
An industry standard would reduce the need for individual judgement, ensure minimum sampling requirements for all higher risk locations, and enable the adoption of best practices across the industry. Moreover, as facilities modernize, a standard provides a framework for developing EM programs in line with technological improvements.
A Proposed Best Practice Guidance Document
The authors took on the challenge of developing a harmonized risk-based methodology. Collaborating for four years and facilitated by BioPhorum, we have drawn on many years of collective experience designing EM programs for a wide range of facilities across the biopharmaceutical manufacturing industry. As far as we are aware, this is the first attempt to comprehensively document and propose best practices.
The first version of this guidance document, Environmental monitoring (EM): harmonized risk-based approach to selecting monitoring points and defining monitoring plan, was published in early 2019. It comprises a methodology that addresses the four key questions for the design of an EM performance qualification (EMPQ) program and subsequent routine EM program:
- What are the risk factors to consider?
- How to systematically assess a room with these risk factors?
- How to define risk levels?
- What are the minimum standards for monitoring for different risk levels?
Covered in the guidance document are grade A-D rooms, including conventional aseptic filling lines, RABS, isolators, background rooms, and support rooms.
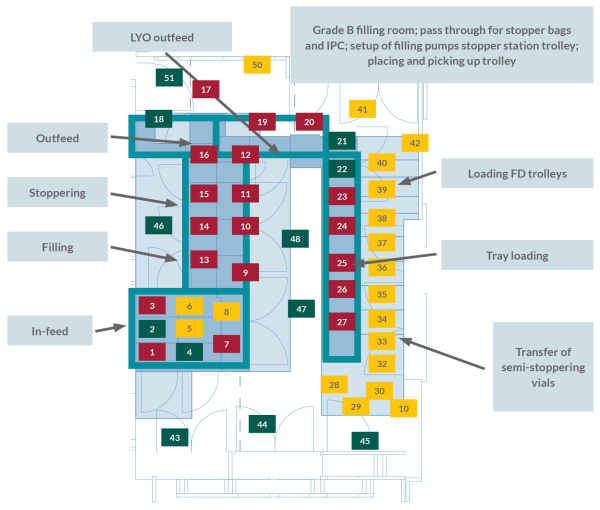
The risk assessment tool systematically evaluates locations within the facility/filling line against standard environmental control elements to determine which areas have the most potential for re-contamination of isolators or contamination risk of rooms during routine manufacturing processes. The six factors identified are:
- Amenability of equipment and surfaces to cleaning and sanitization.
- Personnel presence and flow.
- Material flow.
- Proximity of open product or product contact material.
- The need for interventions/operations by personnel and their complexity.
- The frequency of interventions (only applicable for grade A).
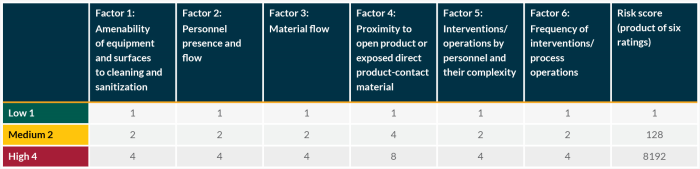
The guidance document describes how to use these six factors to systematically assess all areas of a room and ranks them according to probability of contamination. A set of principles and minimum standards is then proposed to determine where to locate sampling points and select sampling methods to match risk levels.

Next Steps Toward Harmonization Of Best Practices
The guidance has now been updated with a second version. The update to the approach includes revisions in response to feedback and incorporates context from a series of case studies, which we believe adds significant value for all stakeholders in adopting a global standard.
The toolkit is available for free download from the BioPhorum website.
References:
- FDA, “Guidance for industry: Sterile drug products produced by aseptic processing,” Current.
- European Commission, “EU guidelines to good manufacturing practice of medicinal products for human and veterinary use: Annex 1 manufacture of sterile medicinal products,” 2008.
- PDA, “TR13 Vol. 5 (revised): Fundamentals of an environmental monitoring program,” 2014.
- ISO, “14644-2: Cleanrooms and associated controlled environments - Part 2: Specifications for testing and monitoring to prove continued compliance with ISO 14644-1.,” Geneva, 2000.
About The Author:
 Dawn Watson is a director of engineering for Merck & Co. with 26 years of experience in the pharmaceutical industry. She works in the Sterile & Validation Center of Excellence managing a team that leads and executes global initiatives, provides expertise to resolve critical production issues, develops innovative solutions, and ensures network alignment with industry and regulatory expectations.
Dawn Watson is a director of engineering for Merck & Co. with 26 years of experience in the pharmaceutical industry. She works in the Sterile & Validation Center of Excellence managing a team that leads and executes global initiatives, provides expertise to resolve critical production issues, develops innovative solutions, and ensures network alignment with industry and regulatory expectations.