
Environmental Monitoring Performance Qualification In New Drug Manufacturing Facilities
By BioPhorum

Classified environments supporting the manufacture of drug substance and drug product require strict control to minimize the potential for microbiological and particulate contamination of the product. In setting up a new manufacturing facility, a robust process for environmental monitoring (EM) must be established and cleanrooms in the classified areas must be properly qualified.
This article provides an overview of the environmental monitoring performance qualification (EMPQ) process in new facilities, from prerequisites to post-qualification activities.
The Purpose Of EMPQ
Cleanrooms are designed and constructed to meet the requirements of defined classifications in pharmaceutical manufacturing facilities. An EMPQ is an element of testing new cleanrooms after construction and before their use for good manufacturing practice (GMP) manufacturing activities.
The purpose of the EMPQ of cleanrooms is to demonstrate that the cleanroom consistently performs within defined limits and to provide documented evidence that cleanrooms can perform effectively and reproducibly based on predefined parameters. An EMPQ protocol details the frequency, timing, and duration of monitoring over different occupancy states for both microbial and total airborne particle sampling. EMPQ requirements and an EM requalification strategy should be based on a quality risk management system and defined in a validation master plan, contamination control strategy, or equivalent.
Scope
The content below applies to EMPQ of cleanroom environments supporting drug substance and drug product processes. Acceptance criteria for an EMPQ can vary across the industry, so we give guidance on the interpretation of data generated from an EMPQ and what must be assessed as part of the final EMPQ report.
EMPQ In The Facility Life Cycle
An EMPQ is required in all newly constructed facilities/cleanrooms (Grades A to D) where pharmaceutical manufacturing is performed. The requirements and controls are defined by health authorities and demonstrate that the facility is operating in a state of control. These facilities/cleanrooms will continue to undergo regular EM during routine manufacturing following the completion of the EMPQ.
EMPQ is critical to the qualification of a cleanroom and takes place at a defined point in the overall cleanroom life cycle. The relative position of an EMPQ within the facility life cycle is demonstrated in Figure 1.
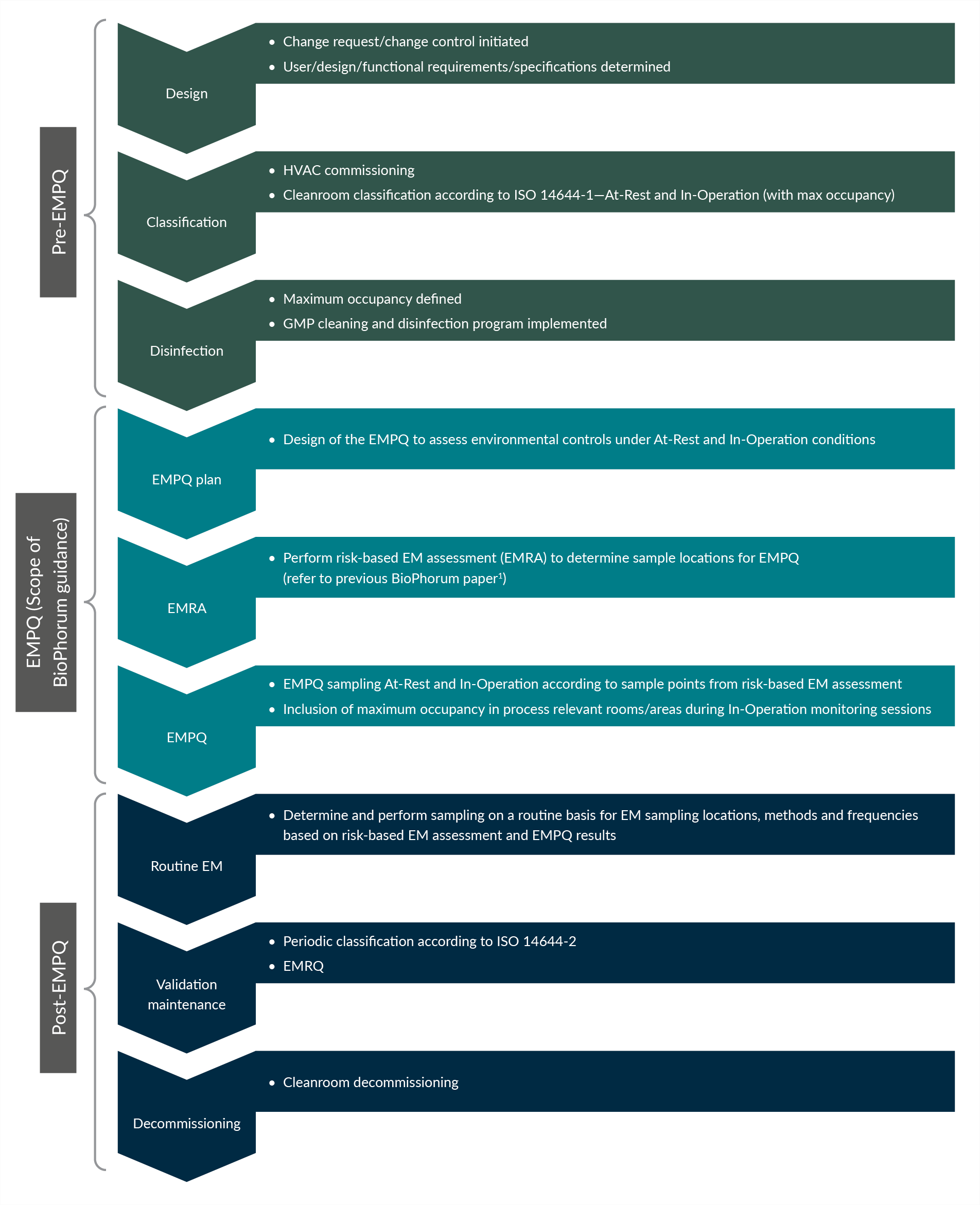
Figure 1: EMPQ within the cleanroom life cycle. Click on figure to enlarge.
Prerequisites To Perform An EMPQ
Before executing an EMPQ, various items must be finalized or defined. Installation qualification and operational qualification must be completed along with any relevant risk assessments. An inexhaustive list of prerequisites for performing an EMPQ, as applicable to the area being qualified, includes:
- manufacturing equipment installation and qualification
- EM equipment installation (where applicable), qualification, and calibration
- maximum occupancy defined.
Establishing Alert Levels And Action Limits
Alert levels and action limits are criteria that indicate whether the control measures are functioning as designed.
Setting Alert Levels
Alert levels are used to provide an early warning of potential drifting from routine operating conditions. For a new facility, historical data and routine operating conditions are not yet established, so the use of alert levels during EMPQ is optional. Opting to use alert levels during EMPQ can provide value in detecting potential adverse trends, such as at a sampling location or within a room, before releasing the area for production.
Setting Action Limits
Action limits are outlined in regulatory guidelines and assigned based on sample type, room classification, and room occupancy state (as applicable). Applying the limits from EU GMP Annex 1 and the FDA Guidance for Industry Sterile Drug Products Produced by Aseptic Processing—Current Good Manufacturing Practice will meet expectations from most other authorities. The EMPQ demonstrates that the facility can be operated within the limits required by regulatory authorities. Tables 1 and 2 are based on EU GMP Annex 1.

Table 1: Maximum permitted total particle concentration for monitoring

Table 2: Maximum action limits for viable particle contamination
EMPQ Sampling Requirements
Room Occupancy States
EMPQ is executed for two room occupancy states (or conditions): at rest and in operation. At rest EMPQ sampling demonstrates that the cleanroom is fit for purpose in the at rest status. In operation EMPQ sampling enables the evaluation of the potential impact that running equipment and the presence of people and processes have on the cleanliness of a cleanroom.
Number Of Sampling Sets
Sampling during EMPQ requires a specific number of sampling sets (also referred to as runs) for each occupancy state. A sampling set is the grouping of one collected sample from each of the EMPQ sampling locations identified for each of the sampling types (e.g., total particle air, microbial surface/swab, active microbial air, passive microbial air). If continuous monitoring (e.g., total particle air, passive air) is used as part of EMPQ, a sampling set can be defined as the set of continuous monitoring samples for the defined operations activity (e.g., engineering batch).
It is recommended that a minimum of one sampling set is collected under at rest conditions and a minimum of three sampling sets are collected under in operation conditions (see Table 3). Any additional EMPQ sampling sets for either occupancy state should be determined by the EM risk-based assessment and process knowledge.

Table 3: Recommended minimum number of sampling sets for EMPQ
Considerations For In Operation EM Sampling
One reason to collect a minimum of three sampling sets under in operation conditions is to allow for sampling during representative time periods (i.e., shifts) and activities related to processing to account for variability in production. When designing the in operation sampling for EMPQ, it is important to consider factors that can impact the state of control and assess whether to incorporate these into the qualification design. The strategy for the design of the in operation sampling should be justified and documented. Factors for consideration when designing an EMPQ include (but are not limited to):
- timing of sampling sets
- in operation activities
- maximum personnel occupancy
- cleaning, disinfection, and decontamination program.
EMPQ Execution
For EMPQ execution, sampling is first completed under at rest conditions, followed by sampling under in operation conditions. Figure 2 is a representative example of EMPQ execution.

Figure 2: Example sequence of EMPQ execution. Click on figure to enlarge.
Acceptance Criteria
EMPQ acceptance criteria must be defined and documented in the EMPQ protocol. Following the completion of sampling, the samples and their results must be assessed against the acceptance criteria to determine if the EMPQ is complete and in compliance. Different acceptance criteria to consider when determining the validity and completeness of the EMPQ include:
- missed samples, invalid samples, and invalid results
- results exceeding the action limit
- results exceeding the alert level
- consecutive results
- recollecting samples.
Final Report Criteria
A final report must document the results and the conclusions of the EMPQ. The criteria required when determining and documenting the validity and completeness of the EMPQ are:
- discussion of results
- protocol deviations
- investigations
- corrective and preventive actions
- confirmation of in operation occupancy state
- overall EMPQ conclusion.
Area Release And Post-Qualification Activities
Once qualification activities are complete, the transition to routine production will require adherence to the criteria documented here.
Before the release of manufacturing systems for GMP use, associated testing must be complete, the impact of deviations/exceptions must be assessed, and a quality review should be performed on the final report to determine the acceptability of the qualification.
At a minimum, the release of manufacturing systems for GMP use should be approved by the quality unit and the EM program owner (where the EM program owner is not the quality unit).
Requalification Strategy
A strategy for periodic requalification must be established and documented outside of the change management process and consider:
- a periodic evaluation of the risk assessment to determine if requalification is necessary
- historical data
- risk-based requalification frequency
- methodologies and sampling locations.
With respect to requalification because of a change management (planned and/or unplanned) or deviation process, this should be commensurate with the level of change and form part of the change management process or quality system as applicable.
Conclusion
Performing an EMPQ is an essential part of the contamination control strategy of each production facility. It is a GMP requirement to qualify cleanrooms over the life cycle of the facility and any planned changes that may impact product quality must be assessed, including the impact on the qualification status.
This article summarizes some of the main points from a recent BioPhorum publication on this topic. To learn more, check out the full paper, Environmental monitoring performance qualification in new facilities: an industry-harmonized approach.
Reference