
EMA Issues Draft Guideline On The Development And Manufacture Of Oligonucleotides
By Peter H. Calcott, Ph.D., FRSC, president and CEO, Calcott Consulting LLC

The EMA issued a draft guidance to industry for comment on July 17, 2024, titled Guideline on the Development and Manufacture of Oligonucleotides.1 The consultation will finish Jan. 31, 2025. This guidance is specific to all the oligonucleotides not covered in other regulations such as Guideline on the Chemistry of Active Substances2 or Chemistry of Active Substances for Veterinary Medicinal Products.3 It also contains requirements and considerations related to conjugation, active substance in solution, medicinal product development, oligonucleotide generics development, oligonucleotide personalized medicine approaches, and clinical trial applications (human products only). However, this guidance does not apply to mRNA products. So, the very well-publicized mRNA Covid vaccines are not covered by this guidance.
The guidance uses the Common Technical Document (CTD) format4 for discussing topics related to active substance (3.2.S), which is logical because the information assembled by the drug sponsor will eventually find its way into the submission (for EU, the MAA). It also offers a separate section pertaining to clinical trial materials. The drug product sections are organized by types of product rather than using the CTD format. Because ICH Q documents were written to exclude synthetic oligonucleotides, this guidance supplements the information in the various ICH documents. So, this guidance advises readers to review current ICH Q series documents for advice. The agency advises the reader to read these in conjunction with GMPs for human and veterinary drugs and especially Annexes 1 and 2 and other annexes.5 In fact, it quotes 20 other documents that should be used in conjunction with this guidance.6 So, I interpret that to mean that this guidance is really to supplement, emphasize, and clarify to the reader what is expected.
As with the CTD, this guidance is divided into active substance and drug product. I will cover them the same way in this article.
Active Substance
Active substance (3.2.S) is divided into seven sections described sequentially below.
Section 1 – 3.2.S.1. General Information
A major point raised is that the structure should be clearly defined, especially any substitutions that make the chain “not normal.” For small interfering RNA (siRNA), the structure of sense- and antisense strands should be provided, and the place of hybridization of the complementary nucleotides of the sense- and antisense strands should be indicated, as well as any unhybridized overhangs in any of the strands. Counter ions should be indicated. The molecular formula and molecular mass of the active substance must be provided; for siRNA, the sense- and antisense strands must also be provided. If relevant, the secondary and tertiary structure (e.g., in the case of hairpin loops or aptamers) should be visualized. Full chemical structure of side chains and linkers is expected. The remainder of the expectations are not surprising. Since the drug substance can be powder or solution, the counter ion must be clearly stated. Stereochemistry should be discussed as relevant.
Section 2 – 3.2.S.2. Manufacture
Since almost all oligonucleotides are synthesized by solid phase technology, any use of technology other than that needs to be very clearly defined. As is expected, details of manufacturing and controls are needed, as is an adequate description of the process and equipment. Because the sequence is critical, the step by step process should be defined with controls, as well as cleavage and chromatography for purification. Particular emphasis should be placed on measures to prevent internal cross-contamination and, hence, incorrect sequencing. Expected yields should be described.
For siRNA, a description of the annealing process should be given with controls, yields, and other process parameters. Any concentration step should be described. Lyophilization steps should be thoroughly described, together with controls.
All starting materials should be described, with special emphasis on those of human and animal origins as well as those nucleotides of a “non-normal structure.” Emphasis on the quality of these starting materials is critical. Do not omit non-nucleotide raw materials from discussion. Appropriate analytical techniques should be used to assure purity of all these reagents.
Control of manufacture is a critical part of the development to assure quality of the end product. Emphasis on the key critical control steps should be described. Risk analysis is a valuable tool to elucidate the critical nature of the controls. A thorough description of process development should be undertaken to elucidate the optimal control strategy. When development has been completed, the process should be validated to modern standards.
Section 3 – 3.2.S.3. Characterization
The structure of the oligonucleotide is a critical element of the program to demonstrate that you have what you claim. The structure should be confirmed by analytical data; this includes the primary, secondary, and tertiary structure, where relevant. Mass spectrometry and its variants are suitable analytical tools for the structure elucidation of oligonucleotides. There are a variety of techniques discussed, including elemental analysis, NMR, UV, circular dichroism, and IR spectroscopy. Stereochemistry and chirality are important parameters to discuss and characterize, as appropriate.
Usually, no biological assay is required for antisense oligonucleotides (siRNA) since they exert their action by annealing to the sense complementary strand. Aptamers bind to target molecules, and that can be the basis of a biological assay. This indirectly measures the fidelity of the tertiary structure. Other techniques that are more general include hygroscopicity, solubility, isoelectric point, and thermogravimetric analysis (including differential scanning calorimetry [DSC] and thermogravimetric analysis). Morphology may be studied by X-ray diffraction and/or DSC. Table 1 illustrates some details on characterization techniques provided in the guidance.
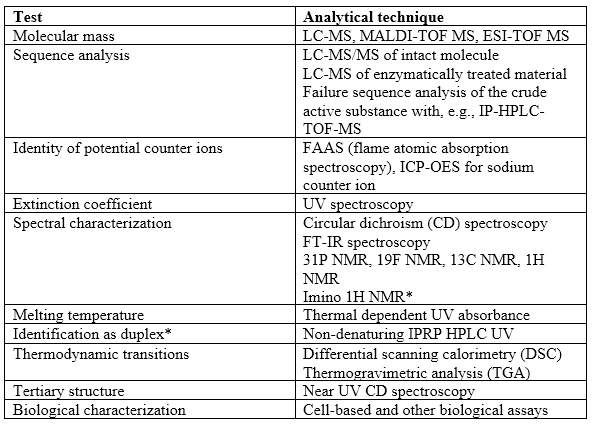
Table 1: Characterization techniques. *Taken from guidance.
Impurities are a critical quality attribute. They can be product or process impurities. The product impurities can be identified by using techniques similar to those used to elucidate and confirm the structure of the product. They can originate from starting materials, the actual process, or from degradation during manufacture. Understanding the source will allow strategies to control and minimize these. There is an expectation to characterize impurities the same as for classic drugs. The guidance describes in detail analysis from all three sources and their implications.
Significant effort should be put into developing assays to measure impurity levels, including defining limit of detection and quantification as appropriate. Some of these might have conflicting biological activities. It may be necessary to develop and perform biological assays to differentiate them. The guidance describes four classes of product impurities dependent on the chemistry involved, which are discussed in detail.
Some impurities are non-nucleotide and are the result of other molecules used in the manufacture. These include reagents, solvents, and leaching from the solid phase material. These should be addressed via purification and/or risk analysis.
Section 4 – 3.2.S.4. Control of Active Substance
Although out of the scope of ICH Q6a,6 it is acceptable to use this guide as a model for developing specifications. Since all these products are administered parenterally, expect to incorporate bioburden and endotoxin into the specification. Table 2 illustrates some pertinent tests for specifications for consideration.
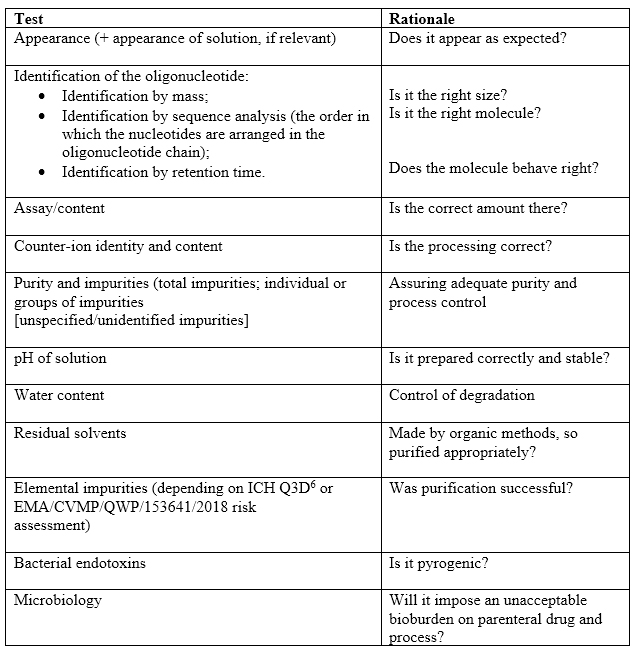
Table 2: Suggested specifications. Taken from guidance with commentary.
Because the sequence of the oligonucleotide is critical for activity, the assay to assure sequence takes on critical importance. Examples of assays are discussed. In the case of double-stranded oligonucleotides, the identity of the duplex and the identity of the single strands should be demonstrated. A combination of non-denaturing and denaturing chromatographical methods in combination with mass spectrometry is generally performed. As an orthogonal method, measurement of the melting point by UV is recommended. For aptamers, sequence confirmation might be challenging, especially due to the lengths and when conjugated, e.g., to PEG. For other assays, good scientific principles and use of Ph. Eur. is advised and encouraged.
For double-stranded oligonucleotides, the purity should be tested both with a non-denaturing method (to allow measurement of single strand residues) and a denaturing method. For aptamers, a test on biological activity should be included in the active substance or finished product specification. Because of the complexity of the molecules and processes, the types of impurities are often complex and difficult to analyze to distinct species, so grouping of impurity types is acceptable.
The quantitation assay should be clearly defined and justified. The level of hydration of powders should be clearly defined. These methods, when developed and operated for a period, should be validated using standard principles of method validation described by ICH.6
When batch analysis is performed and presented, special attention should be paid to the state of the method and the manufacturing process since methods and specifications evolve over the development cycle.
Section 5 – 3.2.S.5. Standards
Appropriate precautions should be taken when dealing with the standard. Since these molecules are hygroscopic, particular care is taken with respect to that parameter. For double stranded oligonucleotides, expect to prepare for both sense- and antisense strands as well as the final product. Complementary strands used as identity tests should be characterized sufficiently.
Section 6 – 3.2.S.6. Container Closure System
The only major point raised is the need to have a moisture-proof container, as the powder forms tend to be hygroscopic. Reactivity may be countered by an inert environment.
Section 7 – 3.2.S.7. Stability
This section describes the major factors. Forced degradation and post commercial commitment to stability are expected, with the stability program paying special attention to the hygroscopicity of powders and the tendency of these molecules to aggregate. ICH Q16 describes elements of the program in general terms.
Drug Products
This section is not broken down by the CTD structure but rather by different types of products.
Conjugations
The use of conjugates to alter the properties of oligonucleotides has emerged. Thus, the nucleotide becomes an intermediate in the manufacture of the agent. Nevertheless, the characterization and control are still critical for the oligonucleotides in this more complex product. Many of the techniques used to characterize the oligonucleotide will apply to the derivative. The details and controls of the process are expected to be documented. Attention should be given to describing and controlling the extent of the reactions and the quantity of unreacted reagents.
Active Substances in Solution
Most oligonucleotides are produced as solids. However, in certain cases, products with excipients in solution have been produced. These pose special challenges because of the potential instability of the resultant solution. Degradation can occur thermally, microbially, and enzymatically. An extensive stability program is expected.
Medicinal Products
The final dosage form should be thoroughly developed, paying special attention to the stability of the final product. Extensive forced degradation studies performed in the active substance stage should address excipients and form of the final product. This development work should support the expected degradation pathways of the drug product. Adequate demonstration of stability of the final product should be presented with data to support the stability, indicating the assays used. Extensive studies of all materials coming into contact with the final product should be undertaken to ensure that the product is not negatively impacted by components used to store the product or the final product.
If the mode of action is due to the primary structure and quantity of material present, then no potency assay is required. But if secondary structure is required for activity, then one is required. Aptamers require a potency assay, which can be performed at either the active substance stage or finished drug stage. If the former, there should be an assurance that the secondary processing does not negatively affect potency. Terminal sterilization is always the preferred method of the EMA, so early on in development its feasibility or non-feasibility should be demonstrated.
Generics Development
Generic versions of oligonucleotides need to adhere to relevant guidelines and regulations such as Guides to Bioequivalence.7 However, complex formulations will require some clinical and/or nonclinical studies. Analytical comparability testing will be useful. In the case of oligonucleotides containing a phosphorothioate linkage, additional investigations regarding the diastereomeric composition should be performed. Suitable state of the art analytical methods should be employed to characterize stereochemistry. Impurity identification and control are paramount and must be detailed. Impurities above 1.0% should be identified and those above 1.5% must be qualified. Product stability must be convincingly demonstrated. It appears that all the characteristics of the product will be examined with equal or more rigorous techniques than conventional biopharmaceuticals.
Clinical Trial Applications
As with any clinical trial application where knowledge is less than complete, patient safety is paramount and every effort to assure it should be taken. Where possible, leverage the new entity from development work performed on similar molecules. Changes should focus on the impact of the changes on the ultimate quality of the product and patient safety. The primary structure should be elucidated, and the secondary structure included as relevant. Impurities should be a major focus.
Personalized Medicines
Personalized medicine is designed for a single patient. If intended for more than a few patients, then the drug loses its personalized nature and flexibility.
For both active substance and finished drug product, suitable state-of-the-art analytical purity methods with stability-indicating properties should be developed. It is not recommended to employ only one method, e.g., IEX or SEC, with limited relevance. Purity testing is central to acceptability.
Stability is still central; omitting stability studies for active substance and drug product and instead relying on public information available for approved medicinal products, without access to the actual data, is not acceptable. A microbiology control strategy should be developed, and sterility of the product applied by parenteral application has to be ensured.
Conclusions
This is a very comprehensive guidance that walks you through your filing for a new oligonucleotide entity. It examines and references a large number of guidances and other regulatory documents that, although were not written for these types of molecules, get you into the mindset for developing and documenting development, characterization, manufacturing, and filing your dossier. Readers are advised to review an FDA guidance on a similar topic published in December 2021.8
References
- Guideline on the Development and Manufacture of Oligonucleotides EMA/ CHMP/CVMP/QWP/262313 /2024
- Guideline on the Chemistry of Active Substances EMA/454576/2016
- Chemistry of Active 58 Substances for Veterinary Medicinal Products EMA/CVMP/QWP/707366/2017
- ICH M4 Common Technical Document. www.ich.org
- EMA Annexes 1 and 2. EU GMP guide Part III.
- 20 Guidances referenced
- Guideline on the Chemistry of Active Substances EMA/454576/2016 and Chemistry of Active Substances for Veterinary Medicinal Products (EMA/CVMP/QWP/707366/2017)
- EU GMP guide Part II: Basic Requirements for Active Substances used as Starting Materials
- EU GMP guide Part II, Q&A 12 on GMP requirements to be applied for the formulation of biological active substances with excipients, when described in the active substance section of a registration dossier
- ICH Q1 A-F Stability testing of new drug substances and drug products – Scientific guidelines (veterinary VICH GL3-5, GL45, GL51 and GL58)
- ICH Q2 Guideline on validation of analytical procedures (veterinary VICH GL1 and GL2)
- ICH Q3A Impurities in new drug substances CPMP/ICH/2737/99 (veterinary VICH GL10)
- ICH Q3B Impurities in new drug products CPMP/ICH/2738/99 (veterinary VICH GL11)
- ICH Q3C Guideline for residual solvents EMA/CHMP/ICH/82260/2006 (veterinary VICH GL18)
- ICH Q3D Elemental impurities EMA/CHMP/ICH/353369/2013 (veterinary Reflection paper 102 EMA/CVMP/QWP/153641/2018)
- ICH Q6A Specifications: Test Procedure and Acceptance Criteria for New Drug Substances and New Drug Products – Chemical Substances CPMP/ICH/367/96 (veterinary VICH GL39)
- ICH Q8 Pharmaceutical development – scientific guideline EMA/CHMP/ICH/167068/2004
- ICH Q9 Quality risk management EMA/CHMP/ICH/24235/2006
- ICH Q11 Guideline on development and manufacture of drug substances (chemical entities and biotechnological/ biological entities) EMA/CHMP/ICH/425213/2011
- ICH Q13 Continuous manufacturing of drug substances and drug products EMA/CHMP/ICH/427817/2021
- ICH M7 Guideline on assessment and control of DNA reactive (mutagenic) impurities in pharmaceuticals to limit potential carcinogenic risk EMA/CHMP/ICH/83812/2013 (veterinary EMA/CVMP/SWP/377245/2016)
- Investigation of Chiral Active Substances 3CC29a for human products, EMEA/CVMP/128/95 for the veterinary products
- Reflection paper on statistical methodology for the comparative assessment of quality attributes in drug development - EMA/CHMP/138502/2017
- CHMP SWP reflection paper on the assessment of the genotoxic potential of antisense oligodeoxynucleotides (EMEA/CHMP/SWP/199726/2004)
- Guideline on the sterilisation of the medicinal product, active substance, excipient and primary container (EMA/CHMP/CVMP/QWP/850374/2015)
- Guideline on the requirements to the chemical and pharmaceutical quality documentation concerning investigational medicinal products in clinical trials - EMA/CHMP/QWP/545525/2017
- Guides to Bioequivalence (CPMP/EWP/QWP/1401/98 Rev.1/ Corr**)
- Draft Guidance - IND Submissions for Individualized Antisense Oligonucleotide Drug Products for Severely Debilitating or Life-Threatening Diseases: Chemistry, Manufacturing, and Controls Recommendations - Guidance for Sponsor-Investigators FDA December 2021
About The Author:
 Peter H. Calcott, D.Phil., is president and CEO of Calcott Consulting LLC, which delivers solutions to pharmaceutical and biotechnology companies in the areas of corporate strategy, supply chain, quality, clinical development, regulatory affairs, corporate compliance, and enterprise e-solutions. He has also served as an expert witness. He also teaches at the University of California, Berkeley in the biotechnology and pharmaceutics postgraduate programs. Previously, he was executive VP at PDL BioPharma, chief quality officer at Chiron and Immunex Corporations, and director of quality assurance for SmithKline Beecham and for Bayer. He has also held positions in R&D, regulatory affairs, process development, and manufacturing at other major pharmaceutical companies. He has successfully licensed products in the biologics, drugs, and device sectors on all six continents. Calcott holds a doctorate in microbial physiology and biochemistry from the University of Sussex in England. He has been a consultant for more than 20 years to government, industry, and academia.
Peter H. Calcott, D.Phil., is president and CEO of Calcott Consulting LLC, which delivers solutions to pharmaceutical and biotechnology companies in the areas of corporate strategy, supply chain, quality, clinical development, regulatory affairs, corporate compliance, and enterprise e-solutions. He has also served as an expert witness. He also teaches at the University of California, Berkeley in the biotechnology and pharmaceutics postgraduate programs. Previously, he was executive VP at PDL BioPharma, chief quality officer at Chiron and Immunex Corporations, and director of quality assurance for SmithKline Beecham and for Bayer. He has also held positions in R&D, regulatory affairs, process development, and manufacturing at other major pharmaceutical companies. He has successfully licensed products in the biologics, drugs, and device sectors on all six continents. Calcott holds a doctorate in microbial physiology and biochemistry from the University of Sussex in England. He has been a consultant for more than 20 years to government, industry, and academia.