
EMA Issues Draft Guideline On Quality Aspects Of mRNA Vaccines
By Minghua Liu, Ph.D., Eliquent Life Sciences

The rapid advancement of mRNA vaccine technology, significantly propelled by its successful application during the COVID-19 pandemic, underscores the importance of clear and robust regulatory frameworks. The EMA's new draft guideline issued in March specifically applies to mRNA vaccines for infectious diseases, including self-amplifying variants, while excluding other mRNA-based medicinal products. It aligns with existing EU regulations and complements other relevant European and ICH guidelines, as well as the European Pharmacopoeia.
This guideline addresses the definition of starting material and provides detailed guidance on manufacturing, characterization, specifications, analytical controls, and regulatory considerations necessary to ensure the consistent quality of mRNA vaccines. It also briefly touches on additional regulatory considerations for strain changes, bi- and multivalent vaccines, and self-amplifying mRNA vaccines. The deadline for public comments is September 30, 2025.
Active Substance Considerations
The guideline defines the mRNA itself as the active substance. Importantly, the guideline identifies starting materials to include nucleotides, the 5' cap or capping reagents, and linear DNA templates.
Linear DNA Template as a Starting Material
Linear DNA receives particular focus. The guideline outlines the need to provide information on its origin, manufacturing process, annotated sequence, and, where applicable, host cell line and cell bank system.
Characterization should address aspects such as pH, DNA concentration, identity and sequence confirmation, poly(A) region integrity, and key impurities like residual DNA, RNA, and proteins. Specifications should reflect process capability and be justified accordingly, supported by relevant stability data. These expectations highlight the importance of robust definition and control of linear DNA as a starting material in mRNA vaccine production.
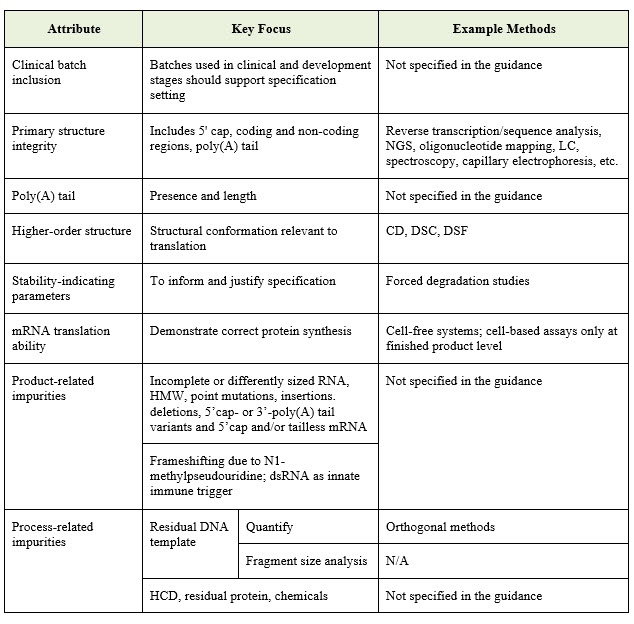
Characterization of the mRNA Active Substance
The guideline specifies a number of key quality attributes and analytical approaches that should be addressed during characterization. These are summarized in the table below:
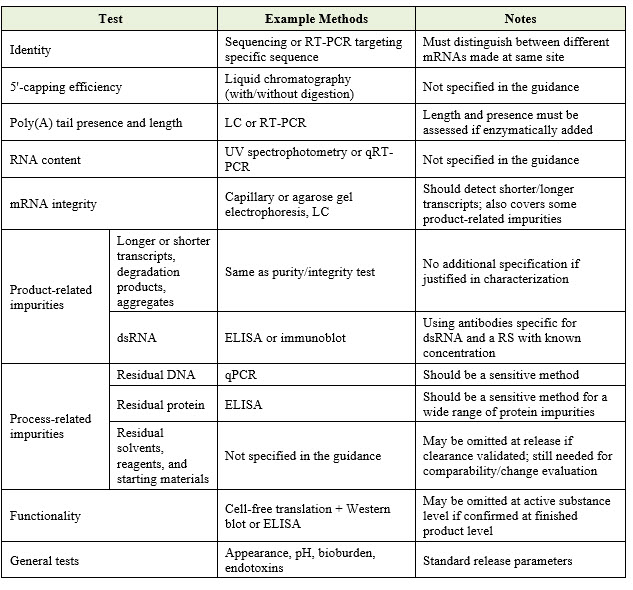
Release Specifications
The guideline sets clear expectations for release testing. Specification limits should be clinically justified in accordance with ICH Q6B. The following table summarizes the key attributes to be controlled and monitored at release of the drug product:
Reference Standards
Where needed, reference standards should be established according to ICH Q2(R2).
Stability
Stability studies should support the proposed shelf life and storage conditions using representative clinical or commercial batches. Key parameters typically include mRNA integrity, poly(A) tail, and RNA content and should reflect any intermediate storage temperatures and freeze/thaw conditions.
Finished Product Considerations
The guideline requires a clear description of the dosage form, container closure system, and the full qualitative and quantitative composition of the finished product, including all lipids and excipients.
Pharmaceutical Development
Pharmaceutical development should be guided by a clearly defined quality target product profile (QTPP), from which critical quality attributes (CQAs) are derived.
The selection of excipients, particularly lipids used for LNP formation, must be justified in terms of safety, functionality, and compatibility with the mRNA. The formulation should be designed to ensure mRNA integrity, effective encapsulation, and delivery.
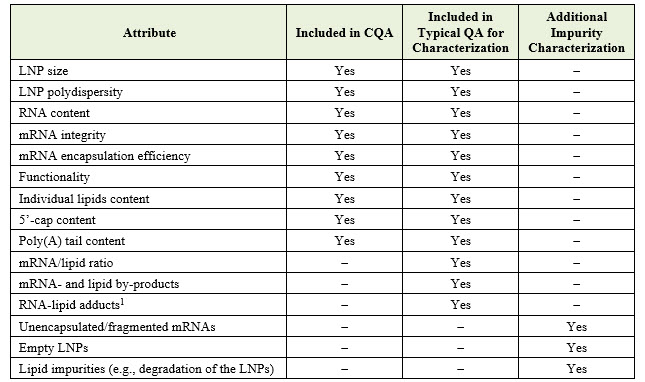
Below is a list of attributes relevant to pharmaceutical development, indicating whether each is identified as a CQA, discussed as a typical quality attribute, and/or mentioned as a product- or process-related impurity requiring characterization:
Manufacture
The manufacturing section should describe any defined intermediates, set appropriate specifications for them, and provide supporting data for their storage conditions and holding times if applicable.
Control of Excipients
Excipients should be compendial when available, with additional quality attributes tested where needed to ensure suitability. If water is a component of the finished drug product, it must meet the quality minimums for water for injection (Ph. Eur.). Excipients of human or animal origin should comply with ICH Q5A and Q11. For novel excipients, full manufacturing, characterization, and control information with appropriate cross-references is expected.
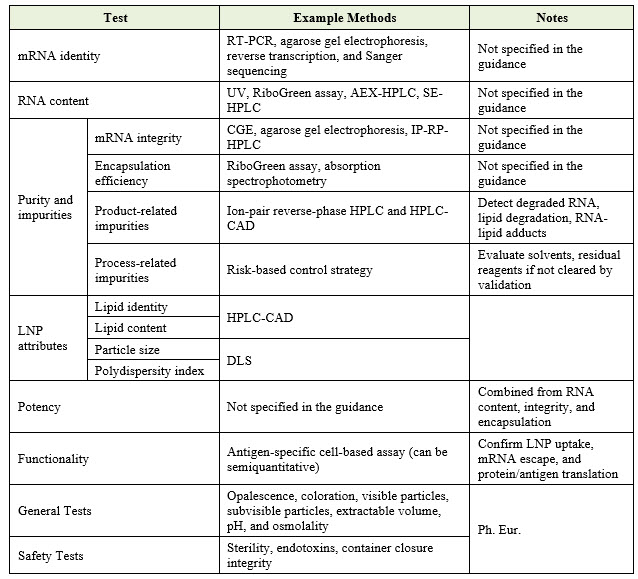
Control of Finished Product
The guideline outlines the critical parameters for finished product testing at release, as summarized below.
Reference Standards
Reference standards for the finished product should be established in line with ICH Q6A, including those for identity, purity, and dsRNA quantification where applicable.
Stability
Stability studies should be conducted in line with ICH Q5C and the ICH’s guideline on Stability Testing of New Drug Substances and Products.
mRNA vaccine-specific parameters should be monitored. In-use stability must be supported if the product is stored under different conditions after thawing (e.g., assessing freeze/thaw effects on integrity). The stability program should include identity, purity, and functionality testing. The proposed storage period should be justified using long-term, real-time, real-condition stability data, including annual testing under all temperatures used for storage. Shelf-life claims should be supported by representative commercial batch data, and prior knowledge from a manufacturing platform may be used if appropriately justified.
Regulatory Considerations
Changes in Existing mRNA Vaccine Strains
When introducing a new strain into an existing mRNA vaccine, the updated specific eCTD sections (with details) expected by the regulatory authority are listed in the guideline. Only changes related to the new strain may be introduced; unrelated parallel changes should not be submitted together.
Bivalent and Multivalent Vaccines
For bivalent and multivalent vaccines, the guideline requires justification of the overall composition, including the specific components of each variant. Based on the manufacturing process, applicants should justify the stage at which testing is conducted, describe critical quality attributes, and provide additional characterization data for the mixture. Methods should enable detection and determination of the ratio of each variant antigen, such as using ddPCR.
Self-amplifying mRNA Vaccines
Self-amplifying mRNA vaccines differ mainly in sequence length and RNA size, while structural features and control strategies remain similar. Additionally, the complete expression profile, including replicase and antigen, together with their functions, should be verified in characterization studies.
Alternative Delivery Systems
The guideline lists cationic nano emulsions, cationic polymers, and cationic peptides as alternative delivery systems. Information on these systems should be included in section 3.2.P.4 (eCTD) if they are part of the drug product composition, or in section 3.2.A.3 (eCTD) if considered novel excipients (including complete description and characterization). The components of such delivery systems should also be included in finished product release testing.
Platform Technology/Prior Knowledge Approach
A product-specific dossier is still required when applying a platform technology-based approach. It is the applicant’s responsibility to provide supporting data, justify the relevance of the prior knowledge, and demonstrate the appropriateness of the proposed strategy. The guideline notes that evaluation should consider factors such as the extent of representative products already licensed or developed, the understanding of product- and platform-specific parameters, and overall risk evaluation, and decisions will be made on a case-by-case basis.
Summary
The EMA draft guideline distills the core quality requirements for mRNA vaccines for infectious disease, covering active substance and finished product considerations, as well as regulatory expectations for strain changes, multivalent formulations, and platform-based approaches. It aims to help developers understand the key principles and prepare robust data packages to support regulatory submissions.
References
- Chromatography and mass spectrometry methods for testing this impurity were mentioned and more details regarding this impurity is specified in the guidance.
About The Author:
 Minghua Liu, Ph.D., is a CMC consultant with extensive regulatory experience at Eliquent Life Sciences. At Eliquent Life Sciences, she has successfully completed over a dozen IND CMC Module 3 submissions and supported BLA filings, specializing in biologics such as mRNA-LNP, ADCs, and cell and gene therapies. Her previous roles were in regulatory compliance and quality management, and she has broad experience working with both domestic and international clients navigating regulatory challenges.
Minghua Liu, Ph.D., is a CMC consultant with extensive regulatory experience at Eliquent Life Sciences. At Eliquent Life Sciences, she has successfully completed over a dozen IND CMC Module 3 submissions and supported BLA filings, specializing in biologics such as mRNA-LNP, ADCs, and cell and gene therapies. Her previous roles were in regulatory compliance and quality management, and she has broad experience working with both domestic and international clients navigating regulatory challenges.