
Drug Facility Design For Live Biotherapeutic Products
By Jimi Kjærsgaard Pettersson, expertise director, NIRAS

Live biotherapeutic products (LBPs) represent a new class of therapeutics using live microorganisms or yeast as active agents to prevent or treat diseases in humans. Unlike traditional probiotics, which are often marketed as dietary supplements for general health, LBPs are developed and regulated as medicinal products with specific clinical indications and rigorous quality, safety, and efficacy requirements, therefore requiring specialized facility designs that balance microbial viability with stringent contamination control.1,2 For pharmaceutical manufacturers entering this emerging field, understanding the unique GMP requirements and engineering solutions becomes critical to ensuring product safety and efficacy while meeting regulatory expectations.
Defining Characteristics Of LBPs
According to the U.S. FDA and the European Pharmacopoeia, an LBP is defined by three key criteria:
- It contains live organisms, such as bacteria or yeast.
- It is intended for the prevention, treatment, or cure of a disease or condition in humans.
- It is not a vaccine, fecal microbiota transplant (FMT), or gene therapy agent.1,3
Mechanisms And Applications
LBPs exert their therapeutic effects through diverse mechanisms, which may include:
- modulating the gut microbiota composition
- regulating immune responses
- producing antimicrobial substances
- enhancing barrier functions within the body after engraftment.3
What Are LBPs And What Can They Be Used For?
The human microbiome, which LBPs are designed to interact with or modify, is a complex ecosystem of microorganisms residing in various body sites. The primary source of LBP strains is the gut microbiome, particularly the colon, which harbors the densest and most diverse microbial populations. However, LBPs also can be derived from and targeted to other microbiome niches, including the skin, oral cavity, and vaginal tract, depending on the intended therapeutic application and disease target.4,6
They are being investigated and developed for a broad spectrum of indications, such as gastrointestinal diseases (e.g., inflammatory bowel disease, recurrent Clostridioides difficile infection), metabolic disorders (e.g., obesity, diabetes), infections, neurological conditions, and certain cancers.5,6
Regulatory Requirements For Facility Design
Regulatory agencies mandate specific facility design requirements for LBP manufacturing to ensure product viability, contamination control, and compliance with GMP. Below are the key requirements based on FDA, EMA, and WHO guidelines.
Containment And Segregation
Regulators require dual containment strategies to:
- prevent cross contamination between strains (critical for spore forming organisms)
- protect products from environmental contaminants.
Key Implementations Include:
- segregated HVAC systems for spore forming vs. non-spore forming strains, with negative pressure cascades (≥15 Pa differential) in spore handling areas
- closed processing systems for anaerobic operations (e.g., nitrogen blanketed milling/encapsulation)
- unidirectional material flow with airlock buffers and three-stage gowning protocols
- modular cleanrooms featuring pre-engineered ISO Class 5–7 environments with epoxy resin surfaces for cleanability and anaerobic condition maintenance.
Cleanroom Design Considerations
Bioprocess cleanrooms for LBPs require specialized features:7
- ISO Class 5 environments (≥0.45 m/s laminar airflow) for critical operations
- laminar airflow with ≥40 air changes/hour in fermentation areas
- negative pressure cascades in spore-forming product suites1
- epoxy resin surfaces with seamless welding for cleanability4
GMP Documentation And Validation
Regulatory submissions must include:
- Master cell bank (MCB) characterization: 16S rRNA sequencing, genomic stability data7
- Process validation: strain-specific parameters for fermentation (pH, temperature, gas mix) and lyophilization cycles
- Stability studies: viability data at 24+ months under ICH Q1A conditions8
Facility Qualifications (IQ/OQ/PQ Protocols):
- Installation qualification: HEPA filter integrity testing, material compatibility checks
- Operational qualification: anaerobic condition maintenance validations
- Performance qualification: microbial retention testing during worst-case simulations
Process Flow Involved In Drug Facility Design For LBPs
The manufacturing process for LBPs is a highly structured and controlled sequence of operations, with each stage carefully designed to preserve the viability of the therapeutic microorganisms while ensuring product quality and regulatory compliance. Facility design plays a crucial role in enabling this process, with dedicated areas and specialized equipment tailored to the unique needs of live microbial production.
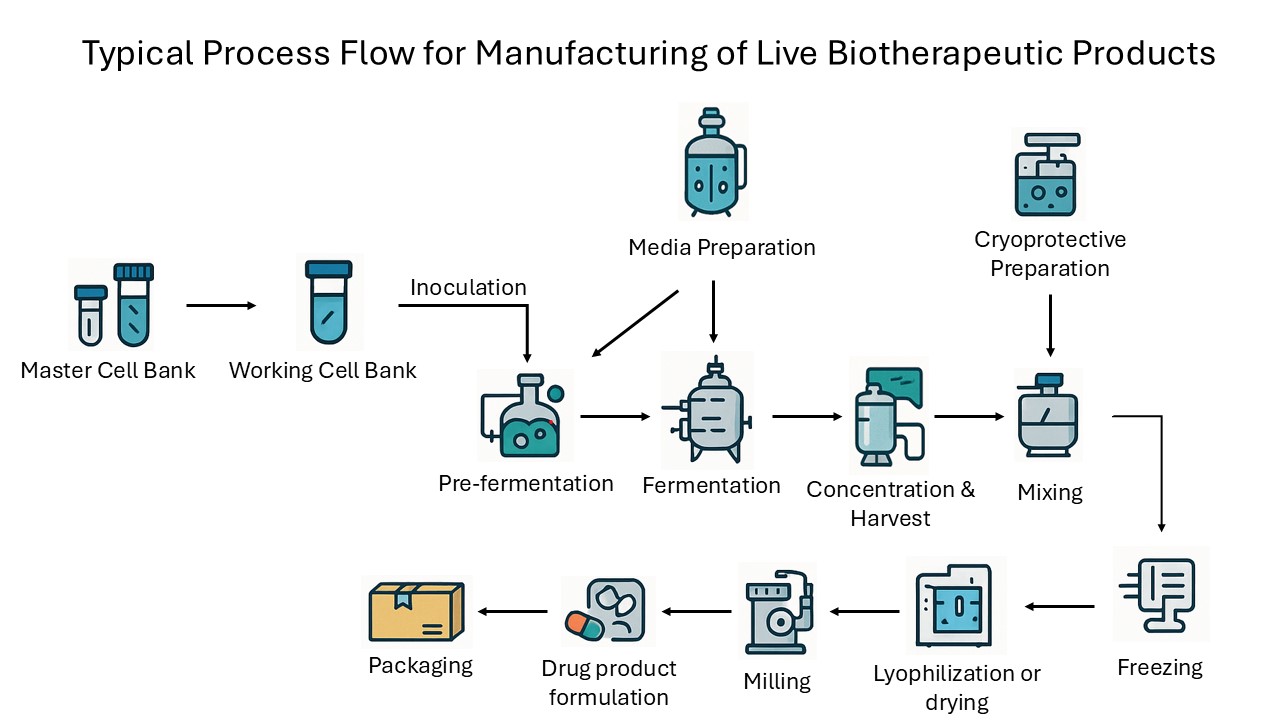
Figure 1: The process flow diagram shows the typical unit operations and process flow involved in the manufacturing of live biotherapeutic products.
The journey begins with the establishment of an MCB, which serves as the foundational source of the production strain. This bank is created in a rigorously controlled environment, where the selected microorganism undergoes comprehensive characterization, including whole genome sequencing, pathogen screening, and phenotypic stability assessments.
Once the MCB is validated, aliquots are used to generate working cell banks (WCBs), which are stored under cryogenic conditions in segregated, contamination-controlled areas. These working banks provide the starting point for each manufacturing campaign, ensuring consistency and traceability from batch to batch.
The production process continues with the preparation of sterile, animal-free growth media. This step takes place in cleanrooms equipped with dedicated vessels and validated sterilization systems to prevent the introduction of contaminants. The prepared media is then inoculated with cells from the WCB in small scale pre-fermentation vessels, where initial microbial growth is closely monitored under controlled temperature, pH, and oxygen conditions.
This pre-fermentation culture is subsequently scaled up in larger fermenters during the fermentation phase, where the microorganisms proliferate under tightly regulated conditions, often anaerobic for gut-derived strains, with continuous monitoring to optimize cell density and viability. Strict oxygen control is crucial in this process to ensure the optimal growth and preservation of anaerobic strains. Fermentation time is highly strain-dependent and can vary significantly based on the specific microbial species and their unique growth characteristics. While some strains may require only one or two days to reach optimal cell density and viability, others may need extended periods of two to three weeks. This variability necessitates precise monitoring and tailored adjustments to maintain the delicate balance of growth conditions, ensuring the microorganisms achieve the desired proliferation while preserving their functional integrity.
Following fermentation, the microbial biomass is separated and concentrated, typically using closed centrifugation or filtration systems that minimize oxygen exposure and the risk of contamination. The concentrated cells are then mixed with a cryoprotective or drying formulation, a critical step designed to protect the microorganisms during subsequent freezing and drying. This formulation, often containing agents such as trehalose or maltodextrin, is prepared and mixed in closed, inert-gas-blanketed systems to maintain anaerobic conditions and ensure homogeneity.
The formulated product is then subjected to controlled freezing, which can be achieved through various methods such as pelletizing, spray freezing, freezing in cryogenic freezers, or freezing performed directly inside the lyophilizer. These approaches ensure rapid and uniform temperature reduction, vital for preserving cell viability.
Once frozen, the product undergoes lyophilization (freeze-drying) or, in some cases, spray drying. These processes remove water from the cells while maintaining their structural and functional integrity. The dried product is then milled to achieve a uniform particle size, using closed milling systems under an inert atmosphere to prevent moisture uptake and oxidative damage.
The next stage involves the formulation of the drug product into its final dosage form, which may be a powder, capsule, or tablet. This is performed in segregated, humidity-controlled rooms, often with nitrogen blanketed encapsulation lines to further protect the live microorganisms from environmental stress. Throughout this stage, strict in-process controls are maintained to ensure content uniformity, microbial viability, and purity.
Finally, the finished product is packaged in aseptic suites designed to prevent post-processing contamination. Packaging materials and processes are selected to maintain product stability, often incorporating cold chain logistics for temperature-sensitive LBPs. Each batch is subjected to rigorous quality control testing, including container closure integrity, labeling verification, and long-term stability studies.
Throughout the entire process, comprehensive GMP documentation is maintained, capturing all critical process parameters (CPPs), in-process controls, critical quality attributes (CQAs), and test results. This meticulous approach ensures that every batch of LBP is produced to the highest standards of safety, efficacy, and regulatory compliance, from the initial cell bank to the final packaged product.

Drug Manufacturing Facility Design
General Design Principles
The following is a suggestion for what a drug manufacturing facility design could look like, adhering to several key principles to ensure safety, efficiency, and product quality.
Zoning is crucial, with clear separation between areas designated for cell banking, formulation, upstream processing, downstream processing, and packaging. A unidirectional flow of materials, personnel, and waste is implemented to prevent cross-contamination. Environmental controls are maintained through ISO-classed cleanrooms, such as ISO 7 and ISO 8, tailored to specific manufacturing stages.
Containment measures align with biosafety levels (BSL1 or BSL2) depending on the microorganism strains used, and inert gas handling systems meet ATEX standards when dealing with flammable materials. Additionally, robust cold chain logistics are implemented to ensure compliance for cryogenic storage and to maintain the integrity of the final product packaging.
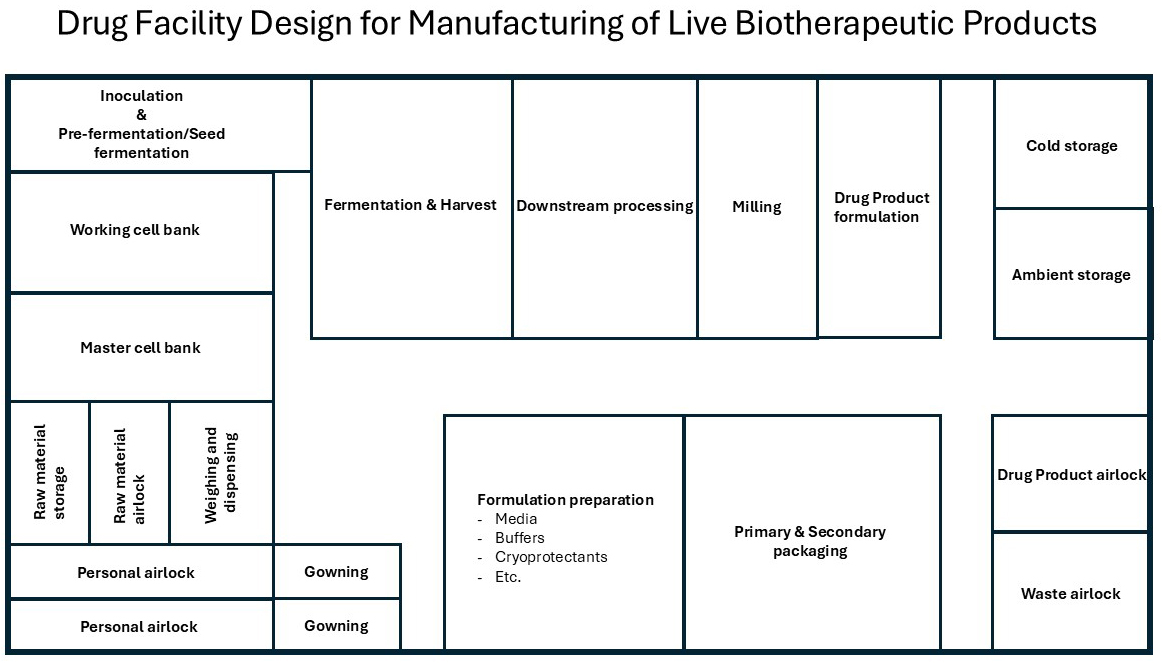
Figure 2: Example of a drug facility design for production of live biotherapeutic product.
Room Layout With Functions
Figure 2 illustrates a conceptual layout for a drug manufacturing facility designed specifically for the production of LBPs. It highlights the spatial organization and functional zoning necessary to maintain safety, efficiency, and product integrity, encompassing areas such as cell banking, upstream processing, and formulation preparation, all adhering to stringent ISO cleanroom classifications and regulatory standards.
The following key processes and facilities are essential for ensuring the smooth and compliant operation of cell bank production and storage:
Cell Bank Production (ISO 7)
MCB production
- Selection and development of the cell line/strain
- Initial characterization and quality control
Cell Banking Storage (CNC)
MCB storage
- Minus 80 degrees C cryogenic freezer (dedicated freezer)
- Genomic QC equipment
WCB storage
- Segregated cryogenic storage (dedicated freezer)
- Identity/viability testing area
Upstream Processing (ISO 7 Cleanroom Suite)
Formulation preparation (media, buffers, cryoprotectants, etc.)
- Sterile mixing tanks (potentially with N₂ blanket)
- Filtration station
- Cryoprotectant mixing tanks (potentially with N₂ blanket)
- Pre-formulation buffer preparation (potentially with N₂ blanket)
Inoculation And Pre-Fermentation (closed processing skids)
- Small seed fermenters
- Temperature/pH/N2/CO2/O₂ controls
Main Fermentation (closed processing skids)
- Large-scale fermenters (single-use or stainless)
- Gas controls (N₂, O2, CO₂)
- Clean-in-place (CIP) access
- Sterile-in-place (SIP)
- Vapor hydrogen peroxide (VHP)
Harvest And Concentration (closed processing skids)
- Centrifuge or tangential flow filtration (TFF)/cross flow filtration (CFF)
- Harvest vessel
Downstream Processing (ISO 7 or 8 depending on exposure)
Drug substance production
Bulk mixing
- Closed system mixer for cryoprotective
- Inert gas (N₂) purge system
Freezing
- Controlled-rate freezers/pelletizer/spray freezer
- Cryogenic safety protocols
Lyophilization
- Freeze dryers with load/unload isolators (for protection against O2 and protection of operator against high potent products)
- Post-dry storage cabinets
Milling
- Nitrogen atmosphere mill
- Contained particle control
Drug Product Formulation (ISO 7 or 8 depending on exposure)
- Encapsulation line, capsule filling or tablet press
- Anaerobic chamber (if needed)
- Humidity control system
- Contained particle control
Packaging And QC (ISO 7)
Primary packaging
- Aseptic filling line
- Cold chain integration (if needed)
Secondary and tertiary packaging and labeling
- Packaging in cartons, bundling, etc.
- Serialization ready labeling
- Cold storage pass through (if needed)
QC labs (placed at another location)
- Microbiology
- Biochemistry
- Environmental monitoring
Support & Logistics
- Personnel airlocks (ISO classed)
- Material airlocks
- Gowning areas – per zone
- Cold storage (intermediate and final)
- minus 80 degrees C, 2 to 8 degrees C, and frozen storage
- Ambient storage (final)
- Waste decontamination room
- Warehouse support
- Utility space
- HVAC, gas lines, UPS systems, clean and black utilities, etc.
References
- Food and Drug Administration, Early Clinical Trials with Live Biotherapeutic Products: Chemistry, Manufacturing, and Control Information, June 2016
- Cordaillat-Simmons, Magali; Rouanet, Alice & Pot, Bruno, Live biotherapeutic products: the importance of a define regulatory framework, 10 September 2020
- Heavy, Mairead K et al., Discovery and delivery strategies for engineered liver biotherapeutic products, 1 September 2021
- Peyton, Duncan, LIVE BIOTHERAPEUTIC PRODUCTS – Not All Microbiome Approaches Are Created Equal, September 2022
- Synapse, For what indications are Live biotherapeutic products being investigated?, 17 March 2025
- Heonhae, Min et al., Live Biotherapeutic Products for Metabolic for Metabolic Diseases: Development Strategies, Challenges, and Future Directions, 11 March 2025
- Annex 2, WHO good manufacturing practices for biological products
- Barberio, Dana, Navigating regulatory and analytical challenges in live biotherapeutic product development and manufacturing, 15 August 2024
About The Author:
 Jimi Pettersson is expertise director at NIRAS, with over 15 years of experience driving R&D and innovation in life science and pharmaceutical development. He has led the advancement and implementation of novel technologies for live biotherapeutic products, specializing in fermentation, downstream processing, and advanced drying techniques. Pettersson has a strong track record in cross-functional leadership, strategic collaborations with academia and industry, and IP management. He holds an M.Sc. in dairy technology and an Executive MBA from DTU, combining scientific expertise with leadership in the life sciences sector.
Jimi Pettersson is expertise director at NIRAS, with over 15 years of experience driving R&D and innovation in life science and pharmaceutical development. He has led the advancement and implementation of novel technologies for live biotherapeutic products, specializing in fermentation, downstream processing, and advanced drying techniques. Pettersson has a strong track record in cross-functional leadership, strategic collaborations with academia and industry, and IP management. He holds an M.Sc. in dairy technology and an Executive MBA from DTU, combining scientific expertise with leadership in the life sciences sector.