
Developing An Automated GxP Workflow Integrated With 21 CFR Part 11
By Sridhar Sarva, senior general manager, Freyr

Moving from paper to paperless in the pharma and life sciences sector
Regulated industries like pharma and medical devices are responsible for ensuring the quality and efficacy of their products to safeguard the public health. To do so, organizations are required to comply with applicable GxP workflows from the R&D stage until a drug or device reaches the end user. During this product development and compliance journey, organizations need to document and log/record every action as per the defined GxP workflows.
Historically, organizations have used a paper-based approach for documenting and activity logging. However, manually recording the regulated processes and quality systems on paper (in the form of clinical binders, lab notebooks, and batch records) involves possible risks of recording incorrect values, loss of records, or falsification of data, and sometimes it may even be affected by common human nature to miss or forget to complete the record keeping. Thus, manual reporting is a complex, resource-intensive, and time-consuming process that could result in:
- Human errors
- Data integrity issues
- Lack of adequate resources (human resources, time, infrastructure, and equipment)
- Lack of an adequate audit trail
These challenges can be resolved only when there is systematic control (either technological or procedural) over the recording of data throughout the product life cycle. This can be achieved through automated/computerized systems with dynamic process control, data processing, and data record/storage applications. These ensure that all the records are authentic, incorruptible, and (where applicable) secure. Implementing this in a GxP workflow is possible only with full data visibility across the product life cycle. This is considered a good automated manufacturing practice (GAMP).
The electronic records and electronic signatures (ERES) regulations, 21 CFR Part 11 and EU Annex 11, provide life sciences companies an opportunity to reap the organizational benefits of paperless record-keeping systems. ERES provides confidence to organizations and regulators, as the electronic signatures and record controls are not simply a legally binding equivalent to paper-based record keeping and wet ink signatures. They are also considered and proven better than paper-based record keeping in terms of traceability, accountability, and data/record retrieval.
Hence, computer system validation in the pharma and medical device industries should focus on implementing GxP workflows integrated with ERES technical controls in addition to focusing on quality, information security, and data privacy during the software development life cycle (SDLC). The use of validated, effective, and GxP controlled computerized systems enhances the quality assurance of regulated products and associated data/information management. A rationalized, integrated, and balanced validation effort is therefore beneficial. If required, the software providers should provide customized solutions to these regulated industries to achieve the balance.
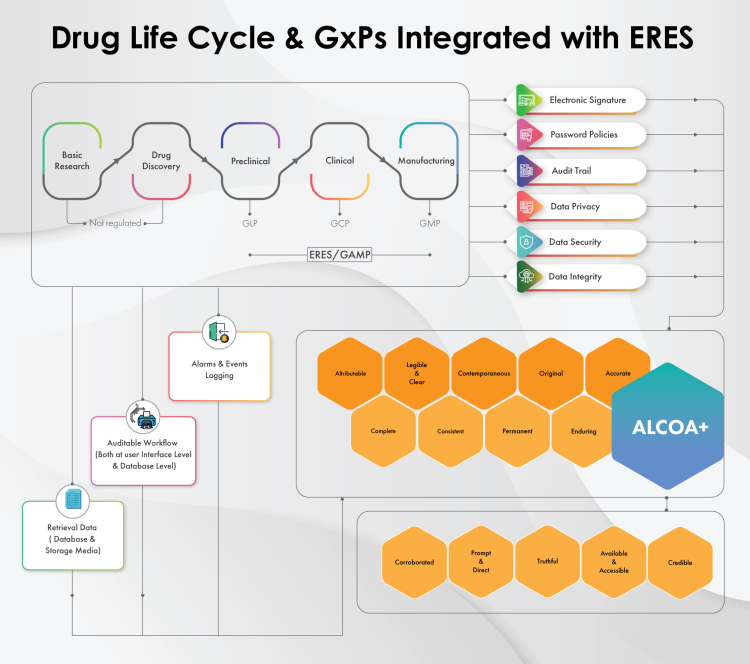
Figure 1: GxP Workflow Integrated with ERES and the Outcome (ALCOA+)
ALCOA+: The Outcome Of Automated GxP Workflow Integrated With ERES
When GxP workflows are integrated with ERES, the associated data will be aligned with certain quality and integrity attributes, known as ALCOA+ (refer to Fig. 1). The ALCOA+ attributes focus on monitoring processes across the data activities to achieve continuous improvements and overall product quality. They ensure that the electronic data management complies with GxPs and drives the data integrity initiatives as follows:
- Attributable: All the data generated during the ERES-integrated GxP workflow are attributable, clearly indicating who (person or system) recorded the data or performed the activity by providing a clear and accurate history of the events.
- Legible: With ERES, the data can be recorded throughout the life cycle, providing uninterrupted accessibility with utmost legibility and clarity. The data can be read and understood even after years and decades.
- Contemporaneous: New entries can be recorded concurrently and instantaneously into the proper GxP record, i.e., recording the activity takes place when the work is performed. Entries in the logbooks are recorded in chronological order and thus can never be backdated.
- Original: The ERES method of documentation preserves the originality of the documents and records them in their unaltered state and generates “certified copies” where required and/or applicable.
- Accurate: All records generated are routinely verified through repetitive calculation or analysis to ensure no scope for errors and thus no scope for changes to the original data. If changes are necessary, those changes are documented with accuracy checks and verification controls that makes referencing them easy.
- Complete: All data, including test results and process re-analysis results, are recorded properly, with all data and metadata.
- Consistent: Documentation and records demonstrate the consistency of required attributes as the flow of data is coherent and performed in the expected sequence.
- Permanent: When integrated with ERES, documents and records are defined as the permanent communication (evidence) that captures work history. They are long-lasting and durable, as they are recorded in validated software systems, including spreadsheets and databases.
- Available: ERES makes documents and records readily available for review, audit, or inspection. With ERES, documents and records are clearly indexed and/or appropriately labeled, facilitating retrievals within a reasonable time.
Overall, the GxP workflow integrated with ERES ensures quality management, as it has the benefits of automated workflow with the level of predefined control as well as auditable and audit-ready electronic data. With immediate access to comprehensive and accurate data, manufacturers can not only meet regulatory compliance but also can identify and address issues faster, avoid recalls, and protect patient safety, thereby reducing operational costs.
While integrating GxP workflow with ERES cannot eliminate all the risks, it can be a good beginning toward developing compliant software. There can be inherent risks to workflows mediated or directed by computerized systems. Due to lack of proper requirements defined in the planning stage, computerized systems can influence the decisions or can interrupt the workflows. These risks depend upon the degree of the system’s configuration that is generating or using the data. Poor system configuration may result in data loss during the transfer of data between systems.
Approach For Developing Compliant Software
Compliant software is that in which the GxP workflows are integrated with appropriate technical controls on ERES, inherent quality, information security, and data privacy. While developing such software, it is recommended that organizations, depending upon their user’s requirements and business prospects, adopt Quality by Design (QbD) and risk-based methodology (RBM) to ensure process simplification and focus on key areas, respectively.
QbD can be achieved by:
- instituting the requirements of GxP workflow, ERES, information security, and data privacy into a common control framework
- considering the software testing steps during the user requirements stage
- referring to the existing data or dashboards of deviations or changes – out of specification (OOS), out of trend (OOT), quality control (QC) outlier, and rejected samples – to derive the requirements to minimize repeated errors and reworks
At each step of the workflow transition, decision-making can become more complex as the process owners must understand, accept, and mitigate the risks at their respective levels, substantiated by their respective electronic signatures.
In addition, as the industry is fast adopting artificial intelligence (AI) and the Internet of Things (IOT), organizations can reap the benefits of automation in GxP workflows, which include minimal human errors, less time, assured data integrity, streamlined audit trails, workflow continuity, paperless record-keeping, and process simplification. While the basis of AI could be historical data, trends, and best practices, it is imperative that the workflows integrated with ERES are developed with controls at appropriate decision-making steps. IOT can be used to automate GxP workflow from the tech-transfer stage to bulk manufacturing and retail manufacturing stages, and from the content to carton stages in the pharma/life sciences and medical device industries.
Conclusion
It is an appropriate time for pharma and medical device manufacturing units, laboratories, and CROs to switch to compliant software that provides end-to-end solutions and replaces or decreases the dependency on paper-based workflows. While adapting to this technological shift, it is important to validate that the software used has built-in capabilities that conform to the ERES methodologies. We believe developing such customized software that meets the current requirements of non-paper-based methodologies could be a potential solution for this era of electronic submissions.
About The Author:
 Sridhar Sarva is a senior general manager at Freyr. For 19+ years, he has worked as a regulatory and quality professional in the regulatory, pharma, contract research organizations (CROs), and electronic data capture (EDC)/cloud industries. He is responsible for carrying out multifaceted roles through process architecture in managing regulatory, quality, and information security compliance. Sarva is skilled in regulatory labeling; publishing and CMC; regulatory consulting and training; designing common control frameworks for privacy, security, and regulatory compliance; CAPA; handling compliance audits; and other areas. He has working experience with the FDA, EMA, MHLW (Japan), GxP (pharma and medical devices), ICH, GAMP 5, CSV, 21 CFR Part 11, EU Annex 11, and HIPAA regulations/guidelines for the life sciences industries.
Sridhar Sarva is a senior general manager at Freyr. For 19+ years, he has worked as a regulatory and quality professional in the regulatory, pharma, contract research organizations (CROs), and electronic data capture (EDC)/cloud industries. He is responsible for carrying out multifaceted roles through process architecture in managing regulatory, quality, and information security compliance. Sarva is skilled in regulatory labeling; publishing and CMC; regulatory consulting and training; designing common control frameworks for privacy, security, and regulatory compliance; CAPA; handling compliance audits; and other areas. He has working experience with the FDA, EMA, MHLW (Japan), GxP (pharma and medical devices), ICH, GAMP 5, CSV, 21 CFR Part 11, EU Annex 11, and HIPAA regulations/guidelines for the life sciences industries.