
Developing A Process Performance Qualification Master Plan For Gene Therapies
By BioPhorum

While many of the activities required to successfully validate a traditional biologic process, e.g., monoclonal antibodies, also apply to gene therapy (GT) manufacturing processes, some considerations are unique to GT.
This article provides considerations and a framework for activities outlined in a Process Performance Qualification Master Plan (PPQMP) in the GT industry. For sponsors with established quality systems and process validation (PV) experience, this may serve to identify gaps or additional considerations in existing validation templates for GT products. For companies with limited validation experience, it can be a resource for planning initial PPQ campaigns.
The purpose of the PPQMP is to define the approach and scope of the PPQ activities required to establish that the sponsor’s GT manufacturing process and control strategy are robust and allow for the reproducible commercial manufacture of DS and/or final DP that consistently meets its predefined acceptance criteria.
According to the FDA, the PPQ combines the actual facility, utilities, equipment (each now qualified), and the trained personnel with the commercial manufacturing process, control procedures, and components to produce commercial batches. A successful PPQ will confirm the process design and demonstrate that the commercial manufacturing process performs as expected.
The three-stage PV life cycle approach is:
Stage 1: process design — The commercial manufacturing process is defined during this stage based on knowledge gained through development and scale-up activities.
Stage 2: process qualification — The process design is evaluated during this stage to determine capability for reproducible commercial manufacturing.
Stage 3: continued process verification — Ongoing assurance is gained during this stage through regular monitoring of routine production to ensure the process remains in a state of control.
The PPQMP defines the requirements for Stage 2: process qualification.
Process Validation Acceptance Criteria
Process qualification verifies that when the manufacturing-scale process is run at target conditions within a predefined acceptable range and executed under validation protocols in accordance with health authority guidance for bulk PV, it produces product that meets all prescribed validation acceptance criteria for process consistency and product quality.
The acceptance criteria should be identified in each PPQ protocol and include all critical process parameters (CPPs) and their proven acceptable ranges (PARs), all critical material attributes (CMAs) and their specification limits, all in-process controls (IPCs) and their validation acceptance criteria or action limits, and all release specifications. As a limited development data set may be available for GT programs to inform initial IPCs, all IPCs should be included for assessment with their associated validation acceptance criteria and/or action limits, and any excursions should be holistically assessed to determine validation impact.
Process parameters and their acceptable ranges are classified according to the sponsor site’s standard operating procedures (SOPs) and are based on results from process development and characterization studies, risk assessments, and previous manufacturing experience or platform approaches. Process parameters are intended to be controlled to a specified target, unless intentional variation away from the target is specified as a process option, and they are maintained within operating ranges (usually tighter than their PARs) to demonstrate process robustness. Process parameters and their acceptable ranges are defined in the production batch records and PPQ protocols and are documented and categorized in control strategy reports and associated product document references.
The control strategy is mapped to all production batch records, process flows, and validation protocols to enable direct application of the process control designations. Parameter classifications (i.e., critical and noncritical), as well as the process failure mode and effects analysis, should be provided and parameters should be maintained within their predefined acceptable ranges.
Missing data or data outside of an acceptable range defined in the PPQ protocols, as per the control strategy, must be supported in an investigation that follows deviation management quality procedures and is determined not to impact the process performance or product quality.
Validation Strategy And Approach
The basic concepts and PV requirements for traditional biologics also apply to GT manufacturing processes. However, PV for GT can present some unique challenges for PPQ. In general, specific areas to consider for GT processes include the identification of critical quality attributes (CQAs), establishing specifications, and validation of analytical methods. In some cases, small batch sizes can complicate IPC and release-testing strategies due to a limited amount of material for sampling.
For example, sponsors may consider using surrogate materials for some validation activities (i.e., mixing validation) with documented justification and risk assessment mitigations. In addition, plasmid processes (also with documented justification and risk assessments) may not require full validation for the entire process; however, many of the considerations below may apply. The sections that follow include some specific considerations unique to GT for a successful PPQ campaign for both DS and/or DP.
PPQ Prerequisites
Prerequisites for PPQ batch execution should be defined in the individual PPQ protocols. General requirements include but are not limited to:
- an approved control strategy,
- validated equipment and analytical methods,
- the target product profile in place,
- qualification of cell and plasmid banks,
- approved procedures,
- trained personnel, and
- use of qualified equipment.
In addition, identified potential CPPs and CMAs should be reviewed and approved by quality before protocol execution. A process failure mode and effect analysis approach for some aspects of plasmid processes may be applicable with appropriate documentation and risk assessment. It is also important to define roles and responsibilities before PPQ execution. Table 1 provides an example of prerequisite activities for PPQ.
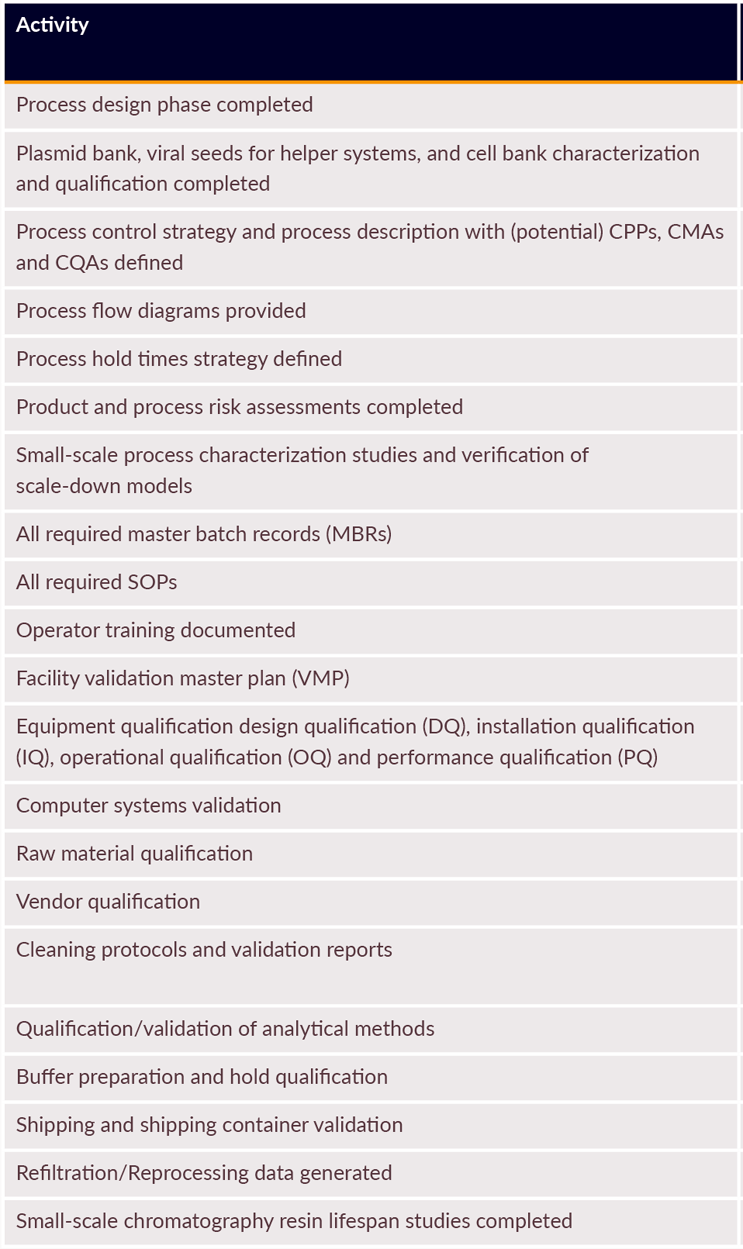
Table 1: Example prerequisite activities for PPQ batch execution
PPQ Strategy
The process understanding achieved through the process design stage should be obtained using a risk-based approach in accordance with quality by design principles and recommendations made in ICH Q8: Pharmaceutical Development and ICH Q99: Quality Risk Management. A risk assessment should be conducted to evaluate unit operations within the manufacturing processes to determine potential high-risk process inputs that require further characterization and gaps in the control strategy that require further mitigation. The risk assessment should be identified and referenced in respective PPQ protocols and reports.
PPQ Approach
The PPQ batches should be performed under PPQ protocols using preapproved master batch records and SOPs representative of the commercial process. The purpose of the manufacturing-scale PPQ batches is to confirm that the manufacturing procedures and control strategy assure the consistency, reproducibility, and quality of the process intermediates at various critical stages in the process, as well as the quality of the final DS and/or DP. This is accomplished by demonstrating that all predefined validation acceptance criteria are met, resulting in product that consistently meets its CQAs.
Drug Substance Process Description
The PPQMP should include a description of the DS manufacturing process as well as a process flow diagram to define the major unit operations. The most common GT approaches use adeno-associated viral (AAV) and/or lentiviral viral vector manufacturing processes.
Drug Product Process Description
If the DP PPQ campaign is separate from DS, the sections in the PPQMP should be adapted to reference the DP manufacturing process. The DP dosage form (e.g., liquid, suspension) and presentation (e.g., vial, syringe, cartridge) as qualified in the manufacturing facility should be provided. The PPQMP should include a description of the DP manufacturing process as well as a process flow diagram to define the major unit operations. The PPQ campaign for DS and DP can be combined under a single PPQMP if the production of both DS and DP is performed continuously (i.e., without frozen storage of the DS). For noncontinuous processes, we recommend that the DS stage is clearly defined and a strategy is applied for appropriate stability and release testing specifications.
Special considerations for this section of your PPQMP:
-
If pooling is planned, consideration should be given to the strategy for residual impurities and these residual impurities should be tested as part of the release of the DS. Extra sampling may be recommended on DS, prior to pooling, and the sponsor should ensure adequate material is available to undertake this testing.
-
In GT processes there may be more variability in the assays used to demonstrate you have reached final concentration compared to other modalities. The sponsor should consider this analytical method variability as they design the approach for demonstrating process robustness.
Reprocessing
Introducing an intermediate or active pharmaceutical ingredient, including one that does not conform to standards or specifications, back into the process and reprocessing by repeating the appropriate unit operation (e.g., refiltration) that is part of the established process, is generally considered acceptable.
Special considerations for this section of your PPQMP:
-
For many GT products, it may not be feasible to reprocess material via operations commonly used for traditional biologics (i.e., by repeating a filtration step) because of the GT product size or sensitivity to shear. Other GT-focused steps may afford reprocessing if they are shown not to impact the product quality (e.g., DNase treatment). The sponsor should perform a risk assessment to determine what process steps may be repeated if required in the process.
Analytical Methods
In-process, release, and compendial assays used for specification testing should be validated before use in validation lots and must be validated before licensure. The requirements for analytical method validation are based on guidance from the International Conference on Harmonization. Appropriate quality control testing should be fit for purpose and identified in individual validation protocols.
Special considerations for this section of your PPQMP:
Relevant guidance for GT analytical procedures can be found in Chemistry, Manufacturing, and Control Information for Human Gene Therapy Investigational New Drug Applications1. Sponsors must ensure scientifically sound principles for assay performance are applied and documented. In summary, sponsors should include:
- detailed description of analytical procedures,
- replication competent virus testing: for non-replicating GT viral vectors, specific testing is recommended due to the potential for these vectors to recombine or revert to a parental or wild-type phenotype at a low frequency, and
- AAV vector production can produce a mixture of full capsids, partly full capsids, and empty capsids. Detection and purification of full capsids from unwanted products is recommended. Assessment of the empty/full capsid ratio is an example where multiple methods are used in the industry.
PPQ Supporting Studies
A number of supporting studies are required as part of validation. Some examples are listed in Table 2.
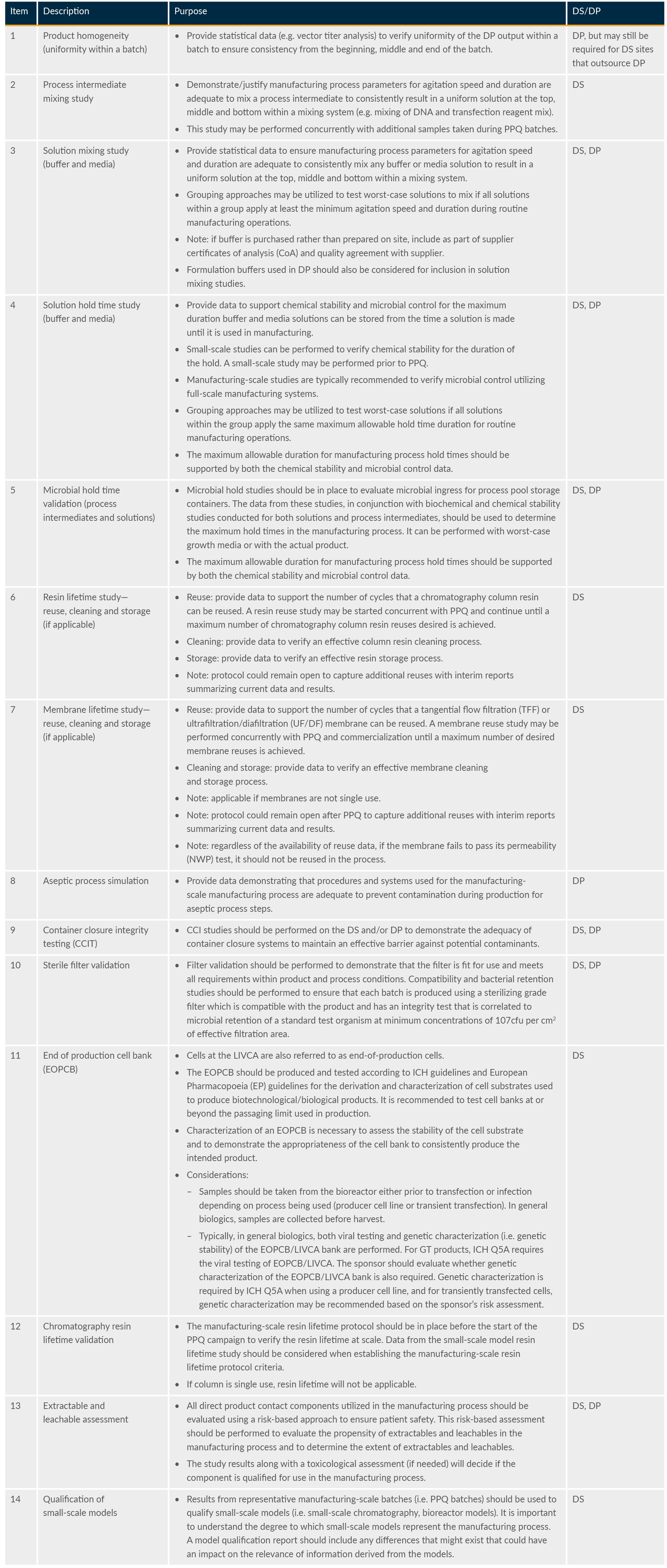
Table 2: PPQ-supportive studies that are required as part of the overall validation package (extract)
Roles And Responsibilities
Roles and responsibilities for PV activities depend on the departmental structure of the sponsoring company and if development and/or manufacturing is performed internally or at a contract development and manufacturing organization.
The manufacturing personnel involved in the execution of the PPQ runs are qualified or trained according to facility-specific procedures and SOPs. Additional training specific to the product PPQ activities is performed and documented according to the site or sponsor SOPs.
Conclusion
Overall, PPQ methodologies established for protein biologics can be leveraged for in vivo GT processes. This is mainly because both modalities employ mammalian cell culture processes and similar downstream processes. This means they require similar validation support studies such as buffer/media and intermediate hold time studies, impurity removal studies, and viral reduction validation studies.
Furthermore, manufacturing equipment is similar across the modalities, which means that, for example, mixing study protocols can be used for both processes.
There are, however, differences that are specific to GT products and production processes that need to be addressed. PPQs typically require extensive, non-routine, in-process sampling and testing. This can be challenging if the process scale and step yields are low, which is currently the case for GT modalities.
Therefore, we advise considering:
- using analytical methods that require small sample volumes, and
- performing validation support studies during clinical manufacturing ahead of PPQ and/or in qualified scale-down models and/or as part of continued process verification.
In addition, the relative immaturity of GT process platforms can cause a higher level of process performance variability. This can be acceptable but must be understood before establishing acceptance criteria for PPQ.
This article summarizes a recent BioPhorum publication on the topic. To read more, check out the full paper in Process performance qualification master plan: considerations for gene therapies.
Reference
1. Chemistry, Manufacturing, and Control (CMC) Information for Human Gene Therapy Investigational New Drug Applications (INDs). Guidance for Industry. FDA CBER January 2020.