
Data Integrity: Surveying The Current Regulatory Landscape
By Barbara Unger, Unger Consulting Inc.
 Failures in data integrity break the essential trust that regulators have with manufactures of medicinal products. Regulatory authorities cannot review all the data that firms generate during the development and commercial lifecycle of every drug. Even during inspections, they review only a small fraction of the data generated. When regulators find that companies have falsified or manipulated electronic or paper data to achieve passing results — or have failed to document and investigate failing results — they lose confidence in all the data presented by the firm, a conclusion with devastating financial consequences.
Failures in data integrity break the essential trust that regulators have with manufactures of medicinal products. Regulatory authorities cannot review all the data that firms generate during the development and commercial lifecycle of every drug. Even during inspections, they review only a small fraction of the data generated. When regulators find that companies have falsified or manipulated electronic or paper data to achieve passing results — or have failed to document and investigate failing results — they lose confidence in all the data presented by the firm, a conclusion with devastating financial consequences.
Data integrity has been the subject of many recent industry trade group meetings involving speakers from the world’s major regulatory authorities, including FDA. The topic, however, is not new and has been the cited in FDA enforcement actions dating back to 1999. The Able Laboratories Form 483 in 2005 created a new level of awareness of data integrity within the industry. Shortly thereafter, the story of Ranbaxy Laboratories and its data integrity deficiencies became the subject of enforcement actions brought about by a whistleblower. And data integrity failures continue to be cited in form 483s and warning letters to this day — with increasing incidence. For companies in India and China, such failures often result in the firm being placed on import alert, which has significant financial ramifications.
In this article, we will review recent regulatory actions related to the highly visible topic of data integrity. First, we will look at data integrity deficiencies cited in FDA warning letters issued between FY 2013 and FY 2015, to sites both inside the U.S. and outside it. Second, we will move to the recently published FDA draft guidance on data integrity, which takes a markedly different approach from the MHRA and WHO guidances, even though FDA has been a leader in enforcement actions in this area for the last 15 years. Finally, we will address the finalized version of the WHO guidance on the topic.
The goal is to provide perspective on the current enforcement environment in the U.S. and abroad. The FDA, WHO, and the EMA have taken enforcement actions on a global basis. Unfortunately, the pharmaceutical industry does not seem to fully appreciate the seriousness with which regulators take these deficiencies, and it has not implemented the corrective and preventive actions necessary to correct these failures.
Data Integrity Deficiencies In FDA Warning Letters
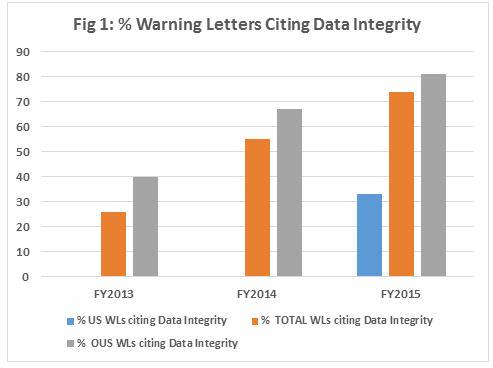
Table 1 shows the number of warning letters issued to firms inside and outside the U.S. (OUS) over the last three FDA fiscal years, excluding those issued to compounding pharmacies. Data integrity deficiencies, including those cited in warning letters, identify the predicate rule(s) to which firms did not adhere — none cite 21 CFR Part 11. Note that even though the total number of warning letters decreased during the time period, the percent that addressed data integrity increased. Figure 1 provides a graphical representation of the data.
Table 1: Data Integrity Deficiencies in FDA Warning Letters (WLs), FY2013-2015
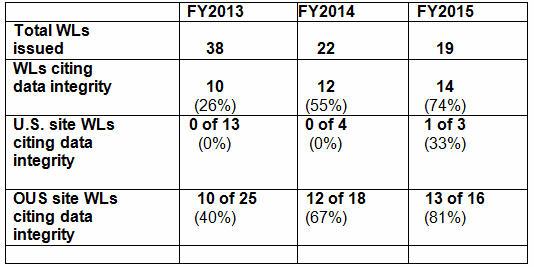
Most data integrity deficiencies addressed in the warning letters focused on the lack of controls over laboratory instrument associated computers/software or failure to contemporaneously record data. The warning letter issued to Sun Pharmaceuticals in early FY2016 (deficiency #6) focuses on manufacturing instrumentation-associated software and computer systems. While related deficiencies have occasionally been identified in past warning letters, the clarity of focus in this deficiency may represent an approach that inspectors will take in moving forward. Watch for more instances of this trend in FY2016, as FDA likely expands its scope to include additional manufacturing floor computer systems.
Several of the warning letters from FY2015 included requirements that approached consent decree-like requirements. Examples may be found in the warning letters issued to: Micro Labs Limited, Apotex Research Private Limited, Hospira Spa, Yunnan Hande Bio-Tech Ltd, and Cadila Healthcare Limited. (For more information on — and links to — these and other FY2013-2015 warning letters, see An Analysis Of Recent CDER Observation & Warning Letter Data.) This text has been refined over the past few years and increased in scope and granularity last year. Currently, the following text appears to be the boiler-plate requirements that FDA includes in instances where serious data integrity deficiencies are identified.
- A comprehensive investigation into the extent of the inaccuracies in data records and reporting. Your investigation should include:
- A detailed investigation protocol and methodology; a summary of all laboratories, manufacturing operations, and systems to be covered by the assessment; and a justification for any part of your operation that you propose to exclude.
- Interviews of current and former employees to identify the nature, scope, and root cause of data inaccuracies. We recommend that these interviews be conducted by a qualified third party.
- An assessment of the extent of data integrity deficiencies at your facility. Identify omissions, alterations, deletions, record destruction, non-contemporaneous record completion, and other deficiencies. Describe all parts of your facility’s operations in which you discovered data integrity lapses.
- A comprehensive retrospective evaluation of the nature of all data integrity deficiencies. We recommend that a qualified third party with specific expertise in the area where potential batches were identified should evaluate all data integrity lapses.
- A current risk assessment of the potential effects of the observed failures on the quality of your drugs. Your assessment should include of the risks to patients caused by the release of drugs affected by a lapse of data integrity, and risks posed by ongoing operations.
- A management strategy for your firm that includes the details of your global corrective action and preventive action plan. Your strategy should include:
- A detailed corrective action plan that describes how you intend to ensure the reliability and completeness of all of the data you generate, including analytical data, manufacturing records, and all data submitted to FDA.
- A comprehensive description of the root causes of your data integrity lapses, including evidence that the scope and depth of the current action plan is commensurate with the findings of the investigation and risk assessment. Indicate whether individuals responsible for data integrity lapses remain able to influence CGMP-related or drug application data at your firm.
- Interim measures describing the actions you have taken or will take to protect patients and to ensure the quality of your drugs, such as notifying your customers, recalling product, conducting additional testing, adding lots to your stability programs to assure stability, drug application actions, and enhanced complaint monitoring.
- Long-term measures describing any remediation efforts and enhancements to procedures, processes, methods, controls, systems, management oversight, and human resources (e.g., training, staffing improvements) designed to ensure the integrity of your company’s data.
- A status report for any of the above activities that are already underway or completed.
Completion of these activities will not happen quickly and will take a concerted effort on the part of the firms involved.
FDA Draft Guidance on Data Integrity
In April 2016, the Federal Register announced availability of the long-awaited 10-page FDA draft guidance on Data Integrity and Compliance with CGMP for comment. For comparison, you can review MHRA GMP Data Integrity Definitions and Guidance for Industry (March 2015) and WHO's Annex 5: Guidance on Good Data and Record Management Practices (May 2016), the latter of which I will explore in more detail below.
The FDA draft guidance is structured in a Q&A format with a total of 18 questions. It focuses heavily on identifying and citing the predicate rules as they apply to electronic records and data integrity, and for this it is an excellent reference. However, it provides little insight into FDA’s intent and actual expectations in this area.
We all read guidance documents to identify regulators’ expectations and actions we might take to ensure compliance. This one, in particular, was anticipated for over two years and addresses FDA’s leadership in enforcement actions over the past 10+ years. Perhaps I had unrealistic expectations, but requirements and expectations in this area can be more easily discerned from a careful reading of warning letter deficiencies and form 483 observations than from reading this draft guidance.
Following are some the areas that I hope are addressed as part of the comment process and revised in the final guidance:
- The guidance fails to address the concept of lifecycle for either computer systems or data. In fact, the term “lifecycle” is not found in the document, even though it is a concept central to the FDA’s guidance on process validation and is also central to associated ICH quality guidelines.
- The guidance does not address an expectation for a risk based data governance process and periodic evaluations of effectiveness of the program to prevent, detect, and remediate data integrity issues. Frequently, FDA warning letters that identify data integrity failures require development of a management strategy to investigate the scope of the shortcoming, including impact on product quality and patient safety, and to address how such failures will be prevented, identified, and remediated in the future. In short, the firm that receives a warning letter must describe a data governance program and a data integrity plan. An example of this requirement is provided at the end of the warning letter recently issued to Emcure Pharmaceuticals.
- Question 16 states that personnel should be trained to detect data integrity issues. While it seems appropriate that all staff should be trained on the concepts and importance of data integrity to ensure product quality and patient safety, it seems excessive and impractical that ALL personnel should be trained to detect data integrity issues. Data reviewers, particularly those that review electronic data, and audit staff should receive special training in the area of detecting data integrity shortcomings. Training for each functional area needs to reflect the roles and responsibilities performed by the staff.
- “FDA invites individuals to report suspected data integrity issues…” and provides an email address to which such communications should be sent. It seems most unusual for FDA to directly solicit what is effectively whistleblower activity in a guidance document. I am not saying this is inappropriate, just that it's unusual.
- With regard to definitions, the guidance does not differentiate between the terms ”back-up” and ”archive” as they relates to electronic records. It would also be ideal if the definitions were harmonized with the two existing guidances.
For now, we await industry and trade group comments, and look forward to the final guidance.
WHO Final Guidance on Good Data and Record Management Practices (Annex 5)
Annex 5 was published as part of the publication of the WHO Technical Report Series No. 996 in May 2016, and represents a finalization of a draft document published for comment in September 2015. WHO initiated work in this area during a meeting in Geneva in April 2014.
Changes from the draft of September 2015 are primarily in reorganization of content and the addition of Appendix I, titled Expectations and examples of special risk management considerations for the implementation of ALCOA (-plus) principles in paper-based and electronic systems. The annex includes the tabulation of information on electronic and paper records, and examples of special risk management considerations that previously were included in a tabulation within the draft guidance. Content has also been expanded in both the appendix and in the document overall, increasing the length of Annex 5 by 10 pages over the 2015 draft.
Following are the changes between the 2015 draft guidance and the finalized 2016 Annex 5. This does not include minor changes in wording or reorganization of the same information. I think it’s impressive that this few changes were made to a document that is 46 pages long.
- Section 2.3 in Aims and Objectives of the Guidance is new.
- The ALCOA-plus entry in the glossary is new.
- The archivist entry in the glossary is new.
- The audit trail entry in the glossary is expanded.
- The control strategy entry in the glossary is new.
- The corrective and preventive action entry in the glossary is new.
- The good data and record management practices entry in the glossary is new and reflects the new term for good documentation practice.
- The quality metrics entry in the glossary is new.
- Section 4.6 Management Governance is expanded.
- Section 4.7 Quality Culture is expanded.
- Section 4.12, providing examples of record keeping methodologies and systems, is expanded.
- Section 4.13, describing durability of data and record media, is new.
- Section 6.4 under Management Governance and Quality Audits is expanded.
- Section 7.2 under Contracted Organizations, Suppliers and Service Providers is expanded.
- Section 7.6 under Contracted Organizations, Suppliers and Service Providers is new. This section addresses expectations where data and document retention is contracted to a third party.
- Sections 11.7 and 11.8 on Data Processing are new.
- Sections 11.15 and 16 on Data Retention and Retrieval are new.
Conclusion
The use of computerized systems in our industry provides great advantages in efficiency and ease of documentation. Failure to correctly configure and validate the systems — along with a small number of firms who have purposefully manipulated the data — has ensured that FDA and other regulatory authorities will continue to focus on this area. It is interesting that the same deficiencies continue to be cited, year after year; industry does not seem to have gotten the message yet. But thanks to specialized meetings held by trade groups like PDA and ISPE, the industry may finally start to gain a better understanding of how essential data integrity is to the manufacture of safe and efficacious products. We need to reach a state where regulators can have confidence in the validity and accuracy of the data on which drugs are approved and released for commercial distribution. In this effort, every member of every pharmaceutical company has an important role to play.
About The Author
Barbara Unger formed Unger Consulting, Inc. in December 2014 to provide GMP auditing and regulatory intelligence services to the pharmaceutical industry. She has extensive expertise in this area having developed, implemented, and maintained the GMP regulatory intelligence program for eight years at Amgen Inc. This included surveillance, analysis, and communication of GMP related legislation, regulations, guidance, and industry compliance enforcement trends. Barbara was the first chairperson of the Rx-360 Monitoring and Reporting work group (2009 to 2014) that summarized and published relevant GMP and supply chain related laws, regulations, and guidance. She also served as the chairperson of the Midwest Discussion Group GMP-Intelligence sub-group from 2010 to 2014.
Before Amgen, Barbara worked for the consulting firm Don Hill and Associates, providing regulatory and quality services to the pharmaceutical industry, and for Eli Lilly and Company in quality and CMC regulatory affairs positions. She began her career in the pharmaceutical / device industry with Hybritech Inc. and received a bachelor's degree in chemistry from the University of Illinois at Urbana-Champaign.