
Contingency Plans: An Essential Quality Management System Risk Tool
By Mark Durivage, Quality Systems Compliance LLC
Probably the biggest concern for anyone implementing, deploying, and maintaining a quality management system  (QMS) is the integration of risk-based thinking. While the concept of risk management is not new, previous practice was more reactionary, primarily focused on detection after the fact, root cause analysis, corrective actions, and preventing recurrence of the failure. Contemporary thinking places the emphasis on considering risks up front (prevention) and having a solid approach to address risk in planning, managing, and driving actions.
(QMS) is the integration of risk-based thinking. While the concept of risk management is not new, previous practice was more reactionary, primarily focused on detection after the fact, root cause analysis, corrective actions, and preventing recurrence of the failure. Contemporary thinking places the emphasis on considering risks up front (prevention) and having a solid approach to address risk in planning, managing, and driving actions.
This article will present the concept of contingency planning and introduce some considerations that can be utilized to develop an effective contingency plan.
Definitions And Background
Several ISO standards, FDA regulations, and international guidance documents provide direction for successfully implementing, maintaining, and sustaining an effective and robust QMS regardless of its type, size, or the products and services it provides. The following requirements speak directly and indirectly about contingency planning.
ISO 9001:2015 Quality management systems — Requirements
4.4.1 The organization shall establish, implement, maintain and continually improve a quality
management system, including the processes needed and their interactions, in accordance with the
requirements of this International Standard.
The organization shall determine the processes needed for the quality management system and their
application throughout the organization, and shall:
d) determine the resources needed for these processes and ensure their availability;
f) address the risks and opportunities as determined in accordance with the requirements of 6.1;
g) evaluate these processes and implement any changes needed to ensure that these processes achieve
their intended results;
6.1.1 When planning for the quality management system, the organization shall consider the issues
referred to in 4.1 and the requirements referred to in 4.2 and determine the risks and opportunities that
need to be addressed to:
a) give assurance that the quality management system can achieve its intended result(s);
b) enhance desirable effects;
c) prevent, or reduce, undesired effects;
d) achieve improvement.
6.1.2 The organization shall plan:
a) actions to address these risks and opportunities;
b) how to:
1) integrate and implement the actions into its quality management system processes (see 4.4);
2) evaluate the effectiveness of these actions.
Actions taken to address risks and opportunities shall be proportionate to the potential impact on the conformity of products and services.
ISO 13485:2016 Medical devices — Quality management systems — Requirements for regulatory purposes
5.4.2 Quality management system planning
Top management shall ensure that:
a) the planning of the quality management system is carried out in order to meet the requirements
given in 4.1, as well as the quality objectives;
b) the integrity of the quality management system is maintained when changes to the quality
management system are planned and implemented.
7.1 Planning of product realization
The organization shall plan and develop the processes needed for product realization. Planning of
product realization shall be consistent with the requirements of the other processes of the quality
management system.
The organization shall document one or more processes for risk management in product realization.
Records of risk management activities shall be maintained (see 4.2.5).
8.5.3 Preventive action
The organization shall determine action to eliminate the causes of potential nonconformities in order
to prevent their occurrence. Preventive actions shall be proportionate to the effects of the potential
problems.
The organization shall document a procedure to describe requirements for:
a) determining potential nonconformities and their causes;
b) evaluating the need for action to prevent occurrence of nonconformities;
c) planning and documenting action needed and implementing such action, including, as appropriate,
updating documentation;
d) verifying that the action does not adversely affect the ability to meet applicable regulatory
requirements or the safety and performance of the medical device;
e) reviewing the effectiveness of the preventive action taken, as appropriate.
Records of the results of any investigations and of action taken shall be maintained (see 4.2.5).
Learn how to apply a risk-based approach to your Process Validation in Mark’s webinar:
How to Establish the Number of Runs Required for Process Validation
21 CFR 211.100 Written procedures; deviations
(a) There shall be written procedures for production and process control designed to assure that the drug products have the identity, strength, quality, and purity they purport or are represented to possess. Such procedures shall include all requirements in this subpart. These written procedures, including any changes, shall be drafted, reviewed, and approved by the appropriate organizational units and reviewed and approved by the quality control unit.
(b) Written production and process control procedures shall be followed in the execution of the various production and process control functions and shall be documented at the time of performance. Any deviation from the written procedures shall be recorded and justified.
21 CFR 820.100 Corrective and preventive action.
(a) Each manufacturer shall establish and maintain procedures for implementing corrective and preventive action. The procedures shall include requirements for:
(1) Analyzing processes, work operations, concessions, quality audit reports, quality records, service records, complaints, returned product, and other sources of quality data to identify existing and potential causes of nonconforming product, or other quality problems. Appropriate statistical methodology shall be employed where necessary to detect recurring quality problems;
(2) Investigating the cause of nonconformities relating to product, processes, and the quality system;
(3) Identifying the action(s) needed to correct and prevent recurrence of nonconforming product and other quality problems;
(4) Verifying or validating the corrective and preventive action to ensure that such action is effective and does not adversely affect the finished device;
(5) Implementing and recording changes in methods and procedures needed to correct and prevent identified quality problems;
(6) Ensuring that information related to quality problems or nonconforming product is disseminated to those directly responsible for assuring the quality of such product or the prevention of such problems; and
(7) Submitting relevant information on identified quality problems, as well as corrective and preventive actions, for management review.
(b) All activities required under this section, and their results, shall be documented.
Identifying The Elements Of A Contingency Plan
One of the best tools to identify which elements should be considered in a contingency plan is the cause-and-effect diagram, better known as the fishbone diagram due to its resemblance to the bones of a fish.
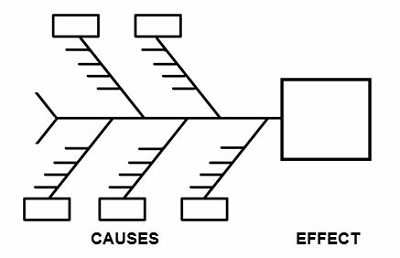
Figure 1: Example cause-and-effect diagram (fishbone diagram)
Brainstorming is a method for generating many creative ideas in a short period of time and can help develop a list of which items should be considered for contingency planning. Brainstorming is conducted by recording ideas about a topic. Each person is asked for an idea in turn, and the session ends when there are no more ideas. The ideas (little horizontal lines) can be generated for each cause (boxes) on the cause-and-effect diagram (see Table 1 for example causes). Once the brainstorming session is completed, an affinity diagram is used to organize ideas into their natural relationships.
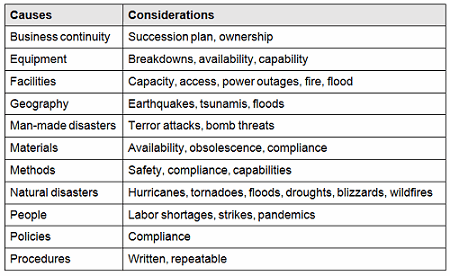
There are many causes for consideration when constructing a cause-and-effect diagram, including business continuity, equipment, facilities, geography, man-made disasters, materials, methods natural disasters, people, policies, procedures, and technology. This list is not all-inclusive and should be based upon an organization’s risk acceptance determination threshold, industry practice, guidance documents, and regulatory requirements.
Table 1: Example Contingency Plan Considerations
With any QMS, there are always many needs and limited resources. One possible method to help determine where the contingency planning resources should be focused is shown in Figure 2. The risk matrix in Figure 2 considers the impact and the likelihood of an occurrence.
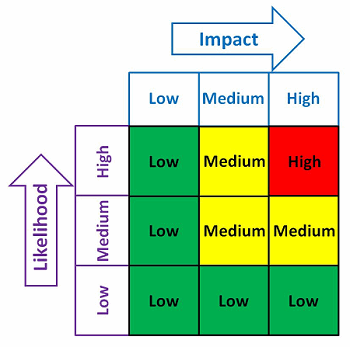
Figure 2: Example risk matrix
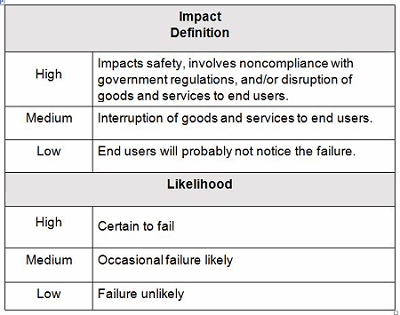
Example impact and likelihood definitions are shown in Table 2. Contingency plans can be developed based upon the indicated level of risk, placing more emphasis and resources on those issues that are high- and medium-risk and less emphasis and resources on those issue that are low-risk.
Table 2: Example Impact And Likelihood Definitions
Contingency Plans
Once the causes and their levels of risk have been determined, it is time to develop the contingency plan. Contingency plans should include a notification and communication plan. They should also include evacuation routes and assembly points where applicable. Contingency plans may indicate the need for backup suppliers, alternative/additional equipment, additional facilities, special insurance, contracts with temporary staffing services, etc. Again, I would like to emphasize that the level of planning and detail should be correlated with the level of risk associated with the event.
Conclusion
The discussion focused on contingency planning to aid the process of identifying, integrating, and mitigating risk throughout the QMS.
I want to reinforce that contingency planning should be based upon an organization’s risk acceptance determination threshold, industry practice, guidance documents, and regulatory requirements.
I cannot emphasize enough the importance documenting the tools and methods used. The requirements and risk management tools presented in this article can and should be utilized based upon industry practice, guidance documents, and regulatory requirements.
This article series has introduced other methods for integrating risk management in the quality management system. The articles in the series include:
- Integrating Risk Management In The Quality Management System — A Primer
- An Introduction To qFMEA – A Tool For QMS Risk Management
- Using Risk-Based Thinking To Manage Suppliers
- Using Trending As A Tool For Risk-Based Thinking
References:
- Durivage, M.A., 2014, Practical Engineering, Process, and Reliability Statistics, Milwaukee, ASQ Quality Press
About The Author:
 Mark Allen Durivage is the managing principal consultant at Quality Systems Compliance LLC and an author of several quality-related books. He earned a B.A.S. in computer aided machining from Siena Heights University and an M.S. in quality management from Eastern Michigan University. Durivage is an ASQ Fellow and holds several ASQ certifications, including CQM/OE, CRE, CQE, CQA, CHA, CBA, CPGP, CSQP, and CSSBB. He also is a Certified Tissue Bank Specialist (CTBS) and holds a Global Regulatory Affairs Certification (RAC). Durivage resides in Lambertville, Michigan. Please feel free to email him at mark.durivage@qscompliance.com with any questions or comments, or connect with him on LinkedIn.
Mark Allen Durivage is the managing principal consultant at Quality Systems Compliance LLC and an author of several quality-related books. He earned a B.A.S. in computer aided machining from Siena Heights University and an M.S. in quality management from Eastern Michigan University. Durivage is an ASQ Fellow and holds several ASQ certifications, including CQM/OE, CRE, CQE, CQA, CHA, CBA, CPGP, CSQP, and CSSBB. He also is a Certified Tissue Bank Specialist (CTBS) and holds a Global Regulatory Affairs Certification (RAC). Durivage resides in Lambertville, Michigan. Please feel free to email him at mark.durivage@qscompliance.com with any questions or comments, or connect with him on LinkedIn.