
Container Closure Integrity Testing Strategies For Gene Therapies
By BioPhorum

Viral vector-based gene therapy drug product (DP) development continues to grow in importance as these therapies demonstrate promising clinical results and gain regulatory approval for the treatment of genetic diseases, different types of cancers, and other indications. The progression of discovery and development of gene therapies is now leading to increased manufacturing capacity to support an increasing number of clinical trials.
However, small batch sizes and high costs associated with gene therapy DP manufacture limit supply and patient access to treatments. Moreover, much of the current regulatory guidance was established based on small molecules and protein therapeutics, causing high material demand for various batch release tests and stability studies.
Container closure integrity, as an important critical quality attribute, is a measurement of the ability of a container closure system to prevent contamination (e.g., microbiological ingress), sustain sterility, and maintain DP stability. Container closure integrity testing (CCIT) has been recognized as having a significant impact on batch yield.
This article provides perspectives on CCIT strategies to minimize the impact on batch yield by examining recent industry survey results as well as best practices. It focuses on CCIT of viral vector gene therapy DP. Cell therapies are out of scope. The CCIT strategy proposed applies to all advanced therapy medicinal product DPs that are manufactured in small lot sizes, typically <500 vials.
Industry Survey Results
Two surveys were undertaken in January 2021 and June 2022 to establish current practices, identify CCIT challenges for gene therapy products, and find potential future approaches to minimize the yield impact of CCIT. A group of 37 subject matter experts from 27 member companies responded to questions across these surveys. Around a quarter of the respondents stated they are focused on development or preclinical phases, about half on clinical early phase, and a quarter on late-phase and commercial programs (see Figure 1).

Figure 1: What phase of development is your product/activity (or your customer's product if a contract organization) currently focused on?
Under regulatory requirements, CCIT is typically performed in place of sterility testing to support the shelf life of DPs rather than release tests for each batch. Some companies may treat CCIT as part of an in-process control or a characterization test for assuring a successful manufacturing process. More than two-thirds of survey respondents indicated that either release, stability, or both were the focus of their CCIT strategy (see Figure 2). Half of respondents intend to implement CCIT in the clinical early phase, a third in the late phase, and the remainder in the development and preclinical phases.
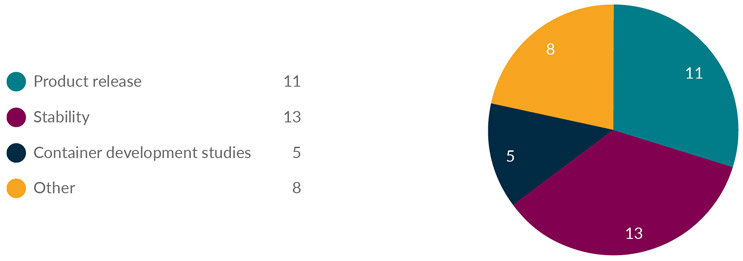
Figure 2: What is the purpose of your CCIT?
It is recommended that a sufficient quantity of containers be sampled at release and each stability time point to produce statistically meaningful results. For certain types of product containers, e.g., glass ampoules, 100% of the batch should be subject to CCIT in accordance with the EU GMP Annex 1 guideline. Visual inspection alone is not considered an acceptable integrity test method. Also, annual CCIT stability testing increases material demand above that already needed for stability study design, further impacting the final yield of the product available for use in clinical trials. This presents a substantial challenge for gene therapy medicines where batch sizes are typically very low (final DP lot sizes for gene therapy products are commonly <500 vials), and the current per unit manufacturing cost of gene therapy DP is substantially higher than protein therapeutics.
In January 2020, the U.S. FDA published Chemistry, Manufacturing, and Control (CMC) Information for Human Gene Therapy Investigational New Drug Applications (INDs). It was recognized in the guidance that it may be challenging or impossible to dedicate a final container or multiple vials for CCIT. Industry was encouraged to consider alternatives to final container testing.
The strategy for CCIT is one area that could be optimized to reduce the sampling and testing overhead from viral vector gene therapy batches. In the initial survey, nine respondents indicated that they performed CCIT on DP, four that they used a placebo, and five responded "other", where this included empty vials (both DP and placebo) or that CCIT has not yet been implemented.
Respondents indicated that a wide variety of CCIT methods are being used and most indicated they intend to perform CCIT on stability at annual time points. Sterility would be performed at the initial and last time points. Most respondents identified that the constraint of small batch sizes is a substantial challenge for implementing CCIT according to current regulatory guidance.
CCIT Methods
Dye ingress and microbial ingress as traditional CCIT methods are probabilistic methods that require a large quantity of containers for method validation and sample testing. In 2016, USP <1207> was implemented to encourage industry to move toward deterministic methods because they produce repeatable and predictable results. Three-quarters of respondents from the survey indicated that they already use or will implement a deterministic method for CCIT (see Figure 3).

Figure 3: Do you plan to test CCIT with deterministic methods?
Table 1 is an extract of the high-level advantages and limitations of each method based on the survey results and literature references.
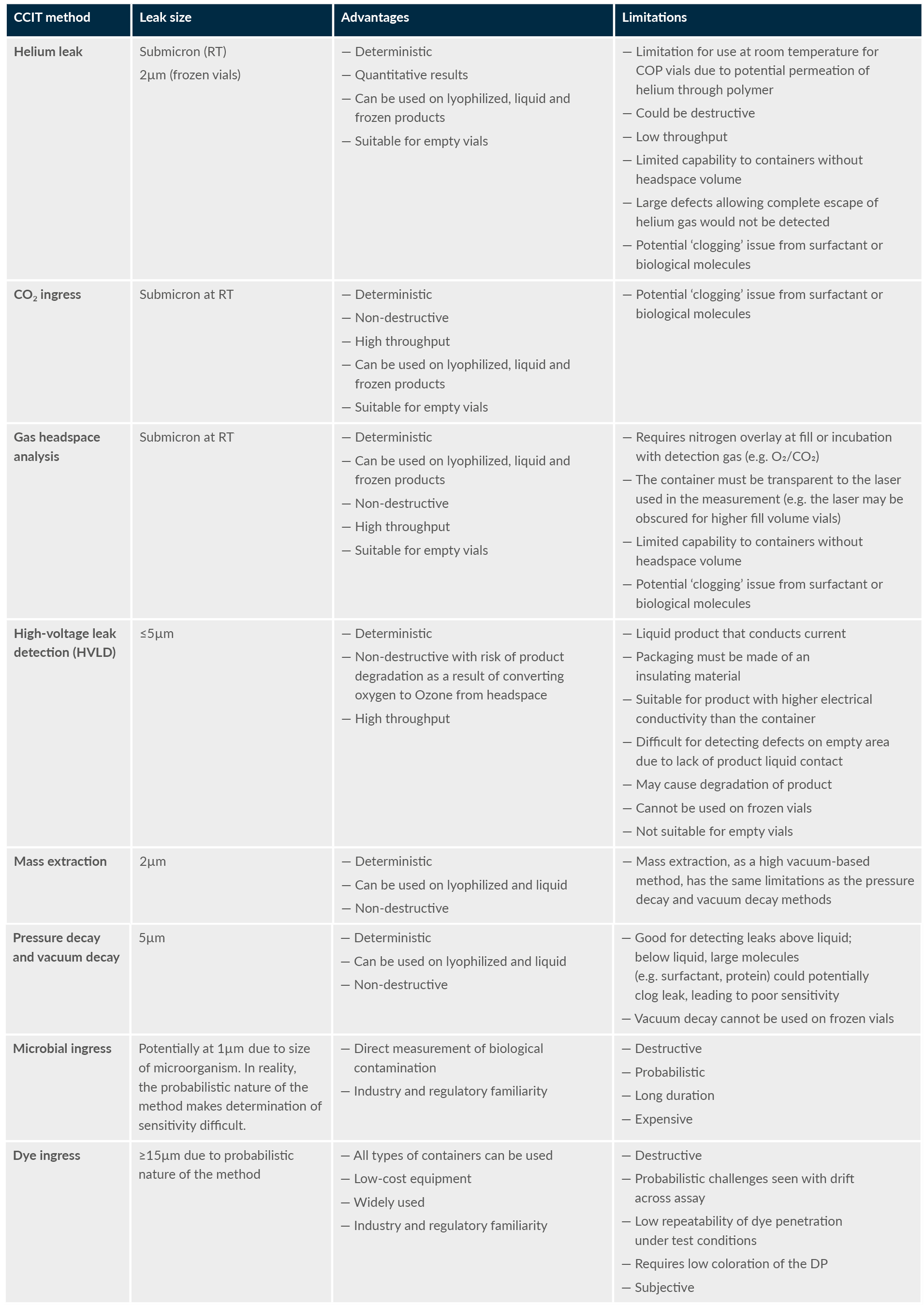
Table 1: Comparison of the advantages and limitations of different CCIT methods (extract)
CCIT Method Validation
As an integral and holistic process, CCIT is evaluated throughout life cycle management, including developing primary packaging, CCIT method validation, and filling line qualification. Many gene therapy products are supplied frozen. Maintaining container integrity is challenging in freezing temperatures, as the low temperature can potentially impact the sealing mechanisms when the vial and rubber stopper have different coefficients of thermal expansion. There may be differences in CCIT when frozen for rubber stoppers and glass vials vs. cyclic olefin polymer vials.
Research shows that leaks that develop during deep cold temperature storage can be resealed when the vials are returned to room temperature, causing false passing CCIT results. Therefore, it is important to assess the suitability of the vial/stopper combination to ensure the degree of shrinkage on the rubber stopper and vial at low temperatures does not fail to maintain container closure integrity. Performing CCIT on frozen vials can directly address this concern.
In general, ICH Q2: Validation of analytical procedures is followed to validate CCIT with key parameters, including accuracy, precision, and detection limit. Drug formulation and containers are not finalized during early development. A change in container during development would require the development and validation of a CCIT method specific to the new container closure system.
Typically, a positive control is generated with a laser-drilled (≥1μm) or microwire-drilled (≥0.2μm) defect of known size to evaluate and verify the detection limit at a 95% confidence interval. In some cases, vials containing a known amount of the detection gas are generated as CCIT defect standards.
CCIT Strategies To Limit Product Loss And Maximize Detection Capability
One approach to limiting the use of product vials for CCIT and sterility testing is to only test a limited number of vials at annual time points. This has a substantial statistical disadvantage but could result in a much-reduced testing overhead of the DP. Sterility testing at annual time points might be an appropriate approach for a very early stage where a CCIT method has not yet been established. However, as sterility testing can also use a substantial number of vials, it would be important to consider stability study strategies that use either CCIT or sterility, but not both.
To minimize product testing overhead losses, CCIT could be achieved using surrogate vials. Such an approach allows larger sample sizes for release and stability testing, leading to a greater capability to detect a CCIT failure while also conserving the product. A reduced testing overhead could help decrease costs and increase access for patients. In this approach, the additional advantage is that surrogate vials do not contain any product that could "plug" (i.e., block) a defect leading to a false passing CCIT.
Empty or water-filled vials do not contain excipients, further reducing the possibility of a false passing CCIT result due to the "plugging" of defects. In terms of minimizing this plugging, empty vials may provide the greatest sensitivity if the selected CCIT method can be applied to empty vials in the same way as filled vials.
Half of survey respondents indicated they would use surrogate vials for CCIT (see Figure 4). For those that will not use the product for CCIT, the impact on the batch yield was a key reason. Most respondents indicated they would generate these vials for CCIT at the end of a DP filling run. The other half of respondents who indicated that they would test CCIT on the DP indicated they would take samples for CCIT at the beginning, middle, and end of the run.

Figure 4: Does your company use placebo/empty/media- or water-filled vials for clinical batch release and/or stability testing for container closure integrity?
Testing of surrogate vials is aligned with principles laid out in the USP <1207.1> and FDA Guidance for Industry: Container and Closure System Integrity Testing in Lieu of Sterility Testing as a Component of the Stability Protocol for Sterile Products, if appropriate justification is provided.
To further reduce method development, validation, and testing overheads, the same method could be used for similar products filled into the same container closure, using a bracketing approach to the method validation. This is aligned with USP <1207> and FDA Guidance for Industry: Container and Closure System Integrity Testing in Lieu of Sterility Testing as a Component of the Stability Protocol for Sterile Products.
Table 2 summarizes the advantages and limitations of different potential approaches to CCIT validation and routine implementation for limited batch-size (i.e., <500 DP vials) gene therapy products. It shows that empty vials have a distinct advantage and fewer limitations overall from a risk-based perspective.
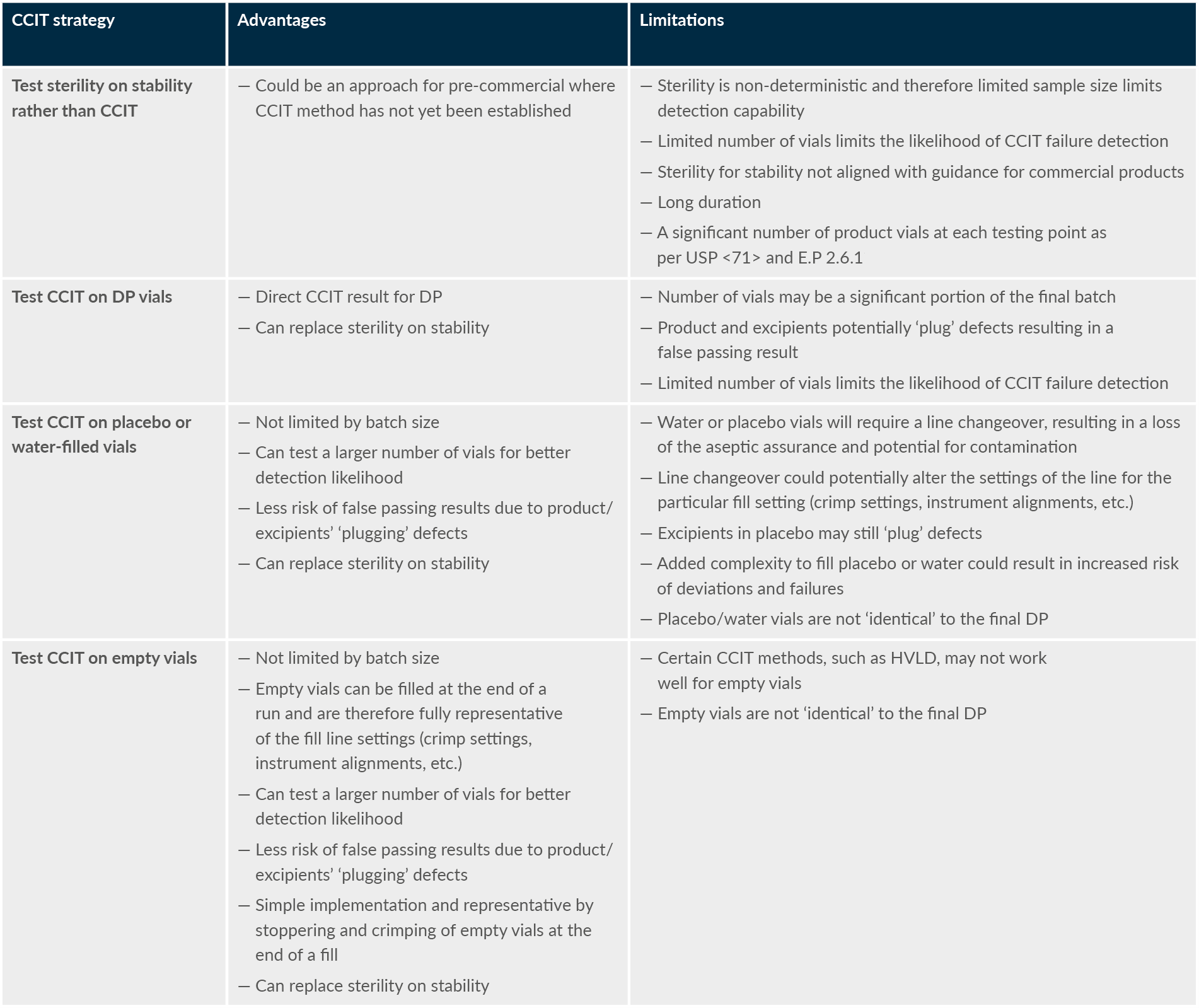
Table 2: Comparison of the advantages and limitations of different CCIT strategies
Conclusion
Productivity and/or total batch sizes, in terms of the number of vials of gene therapy products, are substantial challenges to companies developing these products. With the current state of technology, increasing the scale and/or yield is not a feasible solution to this testing overhead issue, particularly for smaller-scale treatments intended for rare diseases.
A potential approach that could minimize testing overhead losses includes performing CCIT on empty vials, placebo vials, media-filled vials, or water-filled vials (collectively referred to as "surrogate vials") processed with the same equipment. This type of approach may have additional advantages in terms of avoiding false passing results and enabling a greater likelihood of detecting defects.
Furthermore, a larger number of vials can be sampled for CCIT using representative surrogate vials, resulting in an increased likelihood of detecting defects without impacting the batch yield. Method development and validation studies should provide a clear rationale and justification for using a surrogate for CCIT.
This article is a summary of a recent BioPhorum publication on the topic. To read more, check out the full paper in Minimizing the impact of container closure integrity testing on gene therapy batch yield.