
Considerations For Viral Clearance In Cell And Gene Therapy
By BioPhorum

Assuring viral safety in cell and gene therapy (CGT) products poses a unique challenge as the viral vector is a key component of both in vivo and ex vivo gene therapies. Although viral clearance strategies for general biological drug manufacturing and vaccine production will apply to these products, there are unique challenges and considerations for CGT modalities.
Regulatory authorities expect the viral safety and contamination controls for CGT manufacturing to be assured through the application of a comprehensive program of material sourcing, virus testing at appropriate steps of the manufacturing processes, and removal and/or inactivation of adventitious viruses and helper viruses by the manufacturing process.
As with other biological products, such as monoclonal antibodies (mAbs), viral clearance strategies can only be implemented if such processes do not impact product quality. Where viral clearance options are limited, the expectations for virus safety will focus on the testing and control of the raw materials and reagents and on the manufacturing process.
This article evaluates the applicability of the current understanding of viral clearance to the products and processes employed in CGT manufacture to assure the viral safety of these therapies. It focuses on adeno-associated viral (AAV) vectors but also applies to other viral vectors where possible.
CGT products comprise a range of therapeutic modalities, including viral vectors used for gene delivery directly in vivo or ex vivo via gene-modified cell therapy. Cell therapies may be autologous (cells originating from and administered to the same patient) or allogeneic (cells from one donor are modified and expanded to treat multiple patients). While some aspects of the production, characterization, and safety testing for these products are similar to those for recombinant DNA therapies, mAbs, and vaccines, there are unique challenges and considerations for CGT manufacture.
We performed a benchmarking survey and the results indicated that, while most respondents follow International Council for Harmonisation (ICH) Q5A and European Medicines Agency (EMA) guidance, no consensus exists on approaches to viral clearance within the CGT space. Harmonization of approaches toward viral clearance could help sponsors plan studies and reduce the risk of questions from regulatory authorities on viral clearance.
Viral contamination is a potential risk inherent to all biological products manufactured using cells or production cell lines, and CGT is no exception. These rapidly advancing novel modalities carry the risk of endogenous and adventitious viral contamination through the use of cell substrates for manufacturing, the viral vectors used for gene delivery, the environment, and raw materials that may include animal-derived materials. Although the CGT field can leverage the knowledge acquired through experience with biologics manufacturing (including mAbs, recombinant proteins, and viral vaccines), we must account for the unique considerations applicable to specific therapeutic modalities.
Three pillars of viral risk mitigation have been defined in ICH Q5A (R1) Quality of biotechnological products: viral safety evaluation of biotechnology products derived from cell lines of human or animal origin as sourcing, testing, and clearance. These have ensured the safety of biologic products over the past 30 years. These general principles include:
- Careful selection and testing of starting materials.
- Testing in-process materials to ensure they are free of adventitious agents that may be introduced during manufacturing.
- Demonstration that the manufacturing and downstream purification process can clear or inactivate any adventitious and endogenous viruses potentially present to further minimize risk to the final product.
These principles have been found to be effective in preventing any confirmed viral transmittance to patients from recombinant products.
More recently, a draft of the revision of ICH Q5A (R2) was published for industry consultation (with the consultation period ending in February 2023). One of the principal purposes of the revision is to provide industry with guidance on requirements for viral safety and viral clearance for more recent therapeutic modalities that have been evolving over the past decade. These include genetically engineered viral vectors and viral-vectored products, where viral clearance steps do not have a negative impact on the product.
The primary risks of viral contamination in CGT products are associated with the cells used to produce the viral vector or cell therapy, the raw materials used for cell propagation, the cell culture process, and the exposure of the manufacturing process to the environment. Also, viral clearance strategies may be limited due to the nature of the vector and cell therapy products. Inactivation and filtration steps used heavily in the biologics industry to date may not be feasible for these therapies, leaving fewer options to achieve the desired levels of viral clearance, as shown in Figure 1.

Figure 1: Options for viral risk mitigation for CGT products.
Key: AAV – adeno-associated virus; LVV – lentiviral vectors; CTs – cell therapies.
Regulatory Landscape
Viral safety is a major regulatory expectation during the development of biological products, and it can lead to review and approval delays if not adequately addressed. In the U.S., very early in the development of recombinant biological products, the FDA published points to consider and guidelines that outlined recommendations for characterizing cell lines. Initial document listings and adventitious agent testing tables are summarized in references 1 and 2. Other guidance relevant to CGT products can be found in additional points to consider guidelines covering cell lines,3 mAbs,4 and donor selection criteria for allogeneic cell therapy,5 and autologous cell therapy.6 The FDA also published guidelines for industry covering the collection of cells,7,8 cell banking, characterization and release testing, vector construct, and gene delivery systems.9 Microbial vectors were also covered.10 More recently, the FDA published six guidelines relating to CGT, which were revised following industry comments in January 2020. Two of these discuss viral safety.11,12
Key regulatory guidance documents for CGT from the FDA and EMA do not stipulate that viral clearance studies should be performed for CGT products, but they do actively encourage that these studies are performed.11,12,13
Source Of Viral Entry
A critical pillar of a viral safety strategy includes identifying, risk assessing, and addressing any sources of potential viral entry in manufacturing. There are many possible sources of entry, and some examples and considerations are presented in Table 1.
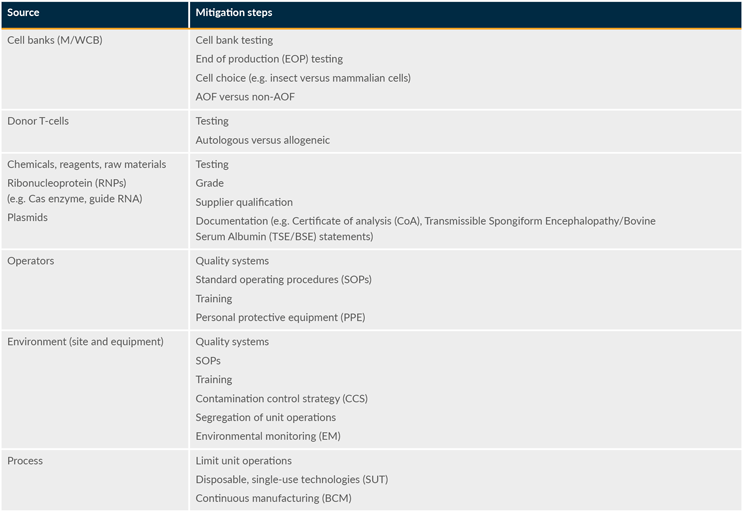
Table 1: Example sources of potential viral entry in CGT. The acronym AOF stands for “animal origin-free.”
Approaches to addressing these risks — including material sourcing, current good manufacturing practice quality systems, documentation, environmental monitoring, and testing — vary widely. Below, we discuss some considerations.
Raw Materials (Adventitious Agents)
Raw material viral safety evaluation and control generally includes documentation, risk assessment, viral safety testing, and supplier qualification. Some general considerations for mitigating contamination from raw materials include:
- Ensure that starting materials such as leukopaks, cell banks, and viral vectors are supported by appropriate documentation (e.g., certificate of analysis or satisfactory test results) and characterization testing to demonstrate compliance or suitability for use.
- Choose the material grade that meets the appropriate quality standards (e.g., compendial) and choose materials free of animal or human origin whenever feasible.
Environment (Adventitious Agents)
Environmental monitoring and control of CGT manufacturing typically focus on mitigating microbial contamination from air, contact surfaces, or operating personnel. Some general considerations include:
- Consider segregating unit operations during facility design to minimize potential cross-contamination. This may include physical walls and differential air pressures (air locks) that control the airflow.
- Ensure a sterile process for critical steps. This may include operation in cleanrooms, laminar flow hoods, or isolators. Maintaining lower particle counts in the air of cleanrooms (e.g., meeting ISO 5 on cleanroom classifications) also reduces the risk of microbial contaminants that can be carried on the particles.
Process Requirements (Vectors Needed For Manufacture)
The process used to manufacture viral vectors poses a unique challenge for CGT products. To produce viral vectors that are unable to replicate independently, helper functions must be provided in any production system. This can be in the form of a helper virus (e.g., Ad5, herpes simplex virus) or plasmids used for transfection or using the helper functions packaged into a baculovirus. In each case, complementing process cells are used that may be mammalian- or insect-derived. When mammalian, especially human, cells are employed for manufacturing (e.g., HEK293, HEK293T, A549, HeLa), additional special considerations for contamination by human viruses are required.
Virus Clearance Validation Strategy
To date, there has been limited detailed guidance for validation of the viral clearance strategy or virus clearance validation. In the draft of the revision of ICH Q5A (R2) and current EMA guidelines, assuring patient safety for conventional mAb processes consists of the comprehensive three-pillar program based on a risk assessment specific to the product in development.
AAV production has borrowed significantly from mAb production best practices. It follows that a three-pillar viral safety strategy (including prevention, detection, and viral clearance studies) would serve as a model for AAV production. The scope for virus clearance validation is based on the type of potential viral contaminants that could be present in the cell source or raw materials (using appropriate testing) and the potential for viral contaminant removal during the downstream purification process. If only potential contamination needs to be considered in the risk assessment, and no endogenous virus-like particles are produced during the cell culture process, a designated clearance target does not exist. However, robust virus clearance is desirable for the process as a whole, ideally including several orthogonal removal steps while considering applicability based on the product’s physicochemical properties. This should be established by evaluating the degree of clearance of various model viruses across different manufacturing unit operations, using spiking studies with the model viruses and representative scale-down models.
Modes Of Viral Clearance
Given that gene therapy products share similar physiochemical properties with wild-type viruses, some of the well-established unit operations developed to target the removal of viral contaminants may not be applicable. For example, parvovirus-retentive fillers (i.e., 20 nm) — developed largely in response to known bioprocessing contaminations with minute virus of mice (MVM) — would be problematic in AAV applications, given that AAV is a member of the Parvoviridae family and very similar in size to MVM. However, recent data indicate that good yields (>90%) can be obtained using larger nanofilters (e.g., 35 nm and 50 nm) with a satisfactory reduction of model virus loads.
Likewise, chemical inactivation of lipid-enveloped viruses, using techniques such as low pH or solvent and detergent holds, would not be compatible with the manufacture of lentiviral vectors and may have differing effects with various AAV serotypes (e.g., pH 3, one hour, may work for one AAV serotype but not for another). In all cases, the proposed removal/inactivation methodologies should be evaluated for the therapeutic viral vector of interest and its manufacturing process.
Conclusion
The CGT field can leverage the knowledge acquired through experience with biologics (including mAbs, recombinant proteins, and viral vaccines) over several decades to develop viral safety strategies for CGT products. It is necessary to account for the unique nature of these products to maintain the potency of the viral vector while removing or inactivating adventitious and/or helper viruses. The need for and extent of virus clearance studies depend on several factors, such as the stage of the clinical trial phase or critical regulatory submissions and the process characterization if a platform technology is used from which virus clearance data could be leveraged.
Our recommendations are based on regulatory and technical understanding in the field. While it is difficult to give a single gold standard approach for a viral clearance validation strategy that will fit all CGT products, this outline will assist in developing a directed approach to address this critical aspect of recombinant AAV manufacture to ensure product safety.
As the industry matures and manufacturing processes continue to develop, a move toward a more platform-based approach may occur. The recent update of ICH Q5 demonstrates an openness of regulatory agencies toward the use of prior knowledge when evaluating viral safety. The knowledge base for mAbs is based on years of study data. A similar body of data is not yet available for CGT. However, as more data are generated in the CGT field, it may be possible to leverage this information to build a more platform-based approach.
This article is a summary of a recent BioPhorum publication on the topic, which includes example viral clearance strategies and the benchmarking survey results. To read more, check out the full paper in Validation: Current approaches and considerations for viral clearance in cell and gene therapy.
References
- Kozak R.W, Durfor C.N., Scribner C.L. Regulatory considerations when developing biological products. Cytotechnology, pp. 203-210. (1992). Regulatory considerations when developing biological products | SpringerLink.
- Kozak, Markus-Sekura CJ and Kozak RW. s.l. In LubinieckiT and Vargo S (eds), Continuous Cell Lines and Contamination testing. Regulatory Practice for Biopharmaceutical Production. Chapter 4;129-187. (1992). : Chernow Ed.Services, Inc. Regulatory Practice for Biopharmaceutical Production | Wiley.
- Points to Consider in the Characterization of Cell Lines used to Produce Biologicals. U.S. Food and Drug Administration. (1993). Points to Consider on the Characterization of Cell Lines Used to Produce Biologicals (fda.gov)
- Points to consider in the manufacture and testing of Monoclonal Antibody Products for Human use. U.S. Food and Drug Administration. (1997). Points to Consider in the Manufacture and Testing of Monoclonal Antibody Products for Human Use | FDA
- Points to consider in the collection, processing and testing of Ex-vivo-activated mononuclear Leukocytes for Administration to humans. U.S. Food and Drug Administration. (1989). Points to Consider Leukocytes for Administration to Humans (fda.gov)
- A proposed approach to the regulation of cellular and Tissue-based Products. U.S. Food and Drug Administration. (1997). Proposed Approach to Regulation of Cellular and Tissue-Based Products | FDA.
- Guideline for industry Eligibility Determination for Donors of Human Cells, Tissue and cellular Tissue-Based Products. U.S. Food and Drug Administration. (2007). Eligibility Determination for Donors of Human Cells, Tissues, and Cellular and Tissue-Based Products | FDA.
- Guideline for industry Considerations for Allogeneic Pancreatic Islet Cell products. U.S. Food and Drug Administration. (2009). Considerations for Allogeneic Pancreatic Islet Cell Products | FDA.
- Guideline for industry Guidance for Human Somatic Cell Therapy and Gene Therapy. March 1998. U.S. Food and Drug Administration. (1998). Guidance for Human Somatic Cell Therapy and Gene Therapy | FDA.
- Guideline for industry Recommendation for Microbial Vectors Used for Gene Therapy. September 2016. U.S. Food and Drug Administration. (2016). Recommendations for Microbial Vectors Used for Gene Therapy | FDA.
- Chemistry, Manufacturing, and Control (CMC) information for Human Gene Therapy Investigational New Drug Applications (January 2020). U.S. Food and Drug Administration. 2020. Chemistry, Manufacturing, and Control (CMC) Information for Human Gene Therapy Investigational New Drug Applications (INDs) | FDA.
- Testing of Retroviral Vector-based Human Gene Therapy Products for Replicative Competent Retrovirus during Product Manufacture and Patient Follow-up. January 2020. U.S. Food and Drug Administration. (2020). Testing of Retroviral Vector-Based Human Gene Therapy Products for Replication Competent Retrovirus During Product Manufacture and Patient Follow-up | FDA.
- EMA/CAT/80183/2014, Guideline on the quality, non-clinical and clinical aspects of gene therapy medicinal products. (22 March 2018). Guideline on the quality, non-clinical and clinical aspects of gene therapy medicinal products (europa.eu).