
Comparing The Top 10 Pharma Inspection Findings From FDA, MHRA, & Russia's Drug Inspectorate

At the FDA/Xavier PharmaLink conference earlier this year, representatives from the Russian Federation State Institute of Drugs and Good Practices (SID&GP), the U.K.’s Medicines and Healthcare products Regulatory Agency (MHRA), and the U.S. FDA shared their top 10 pharma company inspection findings from FY2019, revealing many similarities and a few differences.
A session in which FDA officials have shared and commented on the top 10 483 observations from the previous fiscal year has been a regular annual feature at the FDA/PharmaLink conference (click here for an article covering last year’s session). This year, however, is the first to include findings from the Russian Federation Ministry of Health and the U.K.’s MHRA.
Warning Letter Citations Vs. 483 Observations
The FDA chose to present the top 10 warning letter citations for FY2019 instead of the top 483 observations. Lieutenant Commander Jeffrey Meng, director of the Investigations Branch in the division of Pharmaceutical Quality Operations III within the Office of Regulatory Affairs at the FDA, explained the differences between warning letter citations and 483 observations.
Language in a form 483 states, “this form lists inspectional observations and does not represent a final agency determination regarding your compliance.” On the other hand, an FDA warning letter contains language such as, "this summarizes significant violations of current good manufacturing practices and the failure to properly correct these violations may result in legal action” (see Figure 1).

Figure 1
A form 483 reflects an investigator's best judgment as to what might constitute a violation at the close of the inspection. A warning letter citation reflects the agency's confirmation that the investigator's observation is reportable and a significant violation.
“Although observations are only an investigator's judgment, it does not mean that the observations are not violations of CGMP regulation — they just have not been formally confirmed as such through a somewhat laborious review process,” Meng explained.
In summary, he said, warning letter citations better reflect the agency's current assessment of significant risk across the board. This is clarified in the FDA’s regulatory procedures manual, which states that violations may lead to an enforcement action if the documented violations are not promptly and adequately corrected.
Warning letter observations more closely parallel what was presented by the Russian Federation SID&GP and the U.K.’s MHRA and allow for better comparison.
FDA’s Top 10 Warning Letter Observations For FY2019
Meng presented a slide detailing the FDA’s top 10 warning letter citations for FY2019 (see Figure 2). Each short description is preceded by a reference to the section of U.S. Code of Federal Regulations Title 21 that is being violated.

Figure 2
“To group those top 10 FY19 warning letter citation into themes, I would say one of the major themes was product testing, which includes inadequate stability testing, no finished product testing, as well as no component testing. And this is obviously tied to the quality system,” Meng commented. “Other themes are a lack of data integrity and major equipment failures surrounding cleaning and maintenance.”
Russian Federation State Institute of Drugs and Good Practices’ Top 10
Nadezhda Arkhipova, deputy head of expertise, Department of State Institute of Drugs and Good Practices (SID&GP) of the Ministry of Industry and Trade, lead GMP inspector in the Russian Federation Ministry of Health, presented the top inspection findings with examples and annotations. However, they were not numbered and in some cases were grouped in such a way that related inspection findings were not listed under the same heading. Figure 3 represents what would seem to be a logical grouping and ranking of the findings.

Figure 3
Following are the Russian Federation SID&GP’s top 10 findings with annotations taken from Arkhipova’s slide deck, rearranged to some extent to match the logical grouping and ranking in Figure 3. The text is taken directly from her slides, including words or phrases that were represented in bold.
#1: Laboratory Records Do Not Include Complete Data
No records were kept on the preparation of investigational and reference samples during quality control of a product with a spectrofluorimetric method. No prescribed preparations for spectrofluorometers were recorded, i.e., 30 minutes “heating.” The device is not equipped with a computerized system for recording all activities associated with its use.
No comments on variations in raw data on the results of NDEA and NDMA contents in candesartan API. Overall, there were 31 report versions in Empower 3.
In the chemical lab, logbook records of received raw material samples were written in pencil.
#2: Data Integrity Lapses In the Laboratory
Bioburden testing records do not include data about the media used and about sample preparation.
No real-time records on purified water sampling were made. According to the company’s SOP, the sampling protocol must be printed in advance to be filled in during the sampling. In reality, some protocols were printed and filled in 3 hours later, after the actual sampling.
#3: Lack Of Established Laboratory Controls
Manual integration of chromatograms was used without the proper procedure and double-checking.
The analytical method transfer procedure does not contain step-by-step description and responsibilities of the parties. The whole description of the process fits in a single page instead of the usual at least 10 to 15 pages.
The analytical method description for the API does not specify which exact piece of lab equipment should be used for testing. The company method description contains only general information such as “use a scale,” “use an oven,” etc.
#4: Inadequate Stability Program
Insufficient follow-up on the stability study protocol. For instance, for an oral suspension, one of the required parameters, namely "sedimentation," is omitted with no justification.
Sterile product packed in semi-permeable vials: In 2013, during follow-up stability study of the product, some negative trends were detected, including weight loss and impurities’ concentration growth. In 2015, the company made the decision to reduce the shelf-life of the product from three years to two years. However, in Russia the three-year shelf-life batches were being marketed for sale until 2018. The company failed to inform the Russian regulator about the change.
Regarding the kit of sterile dissolvent (water for injection, or WFI) and freeze-dried product, during a follow-up stability study, multiple out of specification (OOS) results of oxidizable substances were detected in the dissolvent at the points of six, 12, and 24 months. The company informed its local regulator about the OOS and it was decided not to reduce the three-year shelf-life of the dissolvent. The Russian regulator had not been informed about the findings. In Russia, the product was being marketed as it was until the revelation of the Russian inspection.
#5: Inadequate Justification For Storage Conditions
Approved storage conditions for final product (tablets) are up to 30° С. The company provided follow-up stability study conditions within 25 ± 2° С.
#6: Failure To Test Finished Products
There is no quality control of finished product for soft gel capsules. Assay is tested on the intermediate with subsequent calculation for the final dosage form. No justification was provided.
#7: Incorrect Finished Product Testing
Inadequate testing for the presence of E. coli in finished product. Incubation of MacConkey broth is done at 30 to 35° С, instead of 42 to 44° C. Subculture on MacConkey agar is incubated at 20 to 25° С, instead of 30 to 35° C. No 42 to 44° C incubator was presented during the inspection.
#8: Failure To Test Finished Products As Described By Normative Documents
Quality control of finished products for the Russian Federation was not done in line with the normative document (approved specification):
● according to the ND assay, maximum must be 11.00 mg/ampoule; the company spec is 13.75 mg/ampoule
● according to the ND, impurities must be tested. In fact, this control is not done and not included in the company's specification.
#9: Cleaning And Maintenance Of Equipment
Inadequate maintenance was performed on the fluid bed dryer (FBD), i.e., the gasket checkups failed. Powder deposit was found in the technical area. Charging and discharging of FBD are done under negative pressure inside the machine. Drying is done under positive pressure inside. As a result, failed maintenance can lead to cross-contamination (see Figure 4).
Limits for the carryover of product residues are based on OEL (occupational exposure limits) instead of a toxicological evaluation (permitted daily exposure, or PDE).

Figure 4
#10: Manual Cleaning Procedures Not Verified
Manual cleaning procedures of the compactor and the capsule machine have never been verified after their validation. Full cleaning of the machines is done manually.
MHRA’s Top 10 FY2019 Inspection Findings
MHRA GMDP Senior Inspector and GMDP Operations Manager Graham Carroll shared MHRA’s top 10 inspection findings from FY2019 using data his agency had not yet published. “As you can see, it does not vary greatly from what we have heard from my colleagues from the FDA and from Russia,” Carroll remarked (see Figure 5).

Figure 5
Carroll explained that the “reference” column in the table he provided lists the specific references to the EU GMPs similar to the way Meng’s table in Figure 2 references sections of the U.S. Code of Federal Regulations.
To put the inspection findings into context, Carroll presented a column chart showing how many of each citation were discovered that led to its ranking (Figure 6). “Those numbers might look a little bit alarming. The reason for that is really a bit of an artifact of how the data is pulled together. Each deficiency that we cite will almost always have multiple references cited against it, particularly for major and critical deficiencies.”
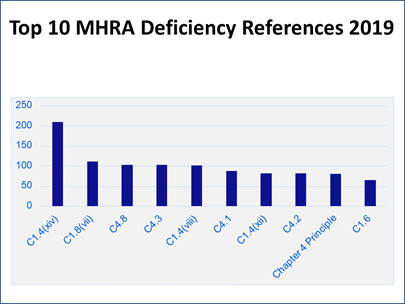
Figure 6
“Let me use a bad cup of coffee as an analogy,” Carroll said. “When we look at why that cup of coffee has turned out to be no good, it turns out that someone has gone and bought really cheap, nasty coffee this week.” In addition, “The water that we used to make that cup of coffee has been left overnight. It has just gone a bit stale. Then we brewed the coffee at far too high of a temperature, so it ended up bitter, and the person who has made it does not really have any training, never drinks coffee, and does not really understand what a good cup of coffee should look like or taste like.
“That is one failure and we would probably report that as one deficiency. But within that, we would have references for the control of raw materials, for the utilities, for the production systems, and for training and personnel issues as well. So, one deficiency might result in four or five hits on this bar chart” (see Figure 6).
Findings From The Three Agencies Have Many Similarities
Common themes across findings from all three agencies include issues with data integrity, deviation investigations, laboratory controls, finished product testing, stability programs, and equipment cleaning and maintenance (Figure 7).
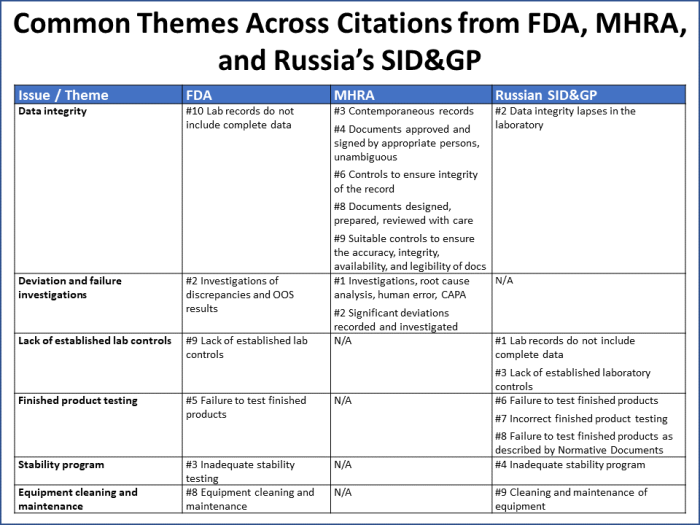
Figure 7
These six issues/themes capture 70% of all the citations reported by the three agencies. It is worth noting that:
- Over half of MHRA’s citations map to data integrity issues.
- FDA uses Q unit citations (#4 and #7) as a catchall for a wide variety of issues that may be cited in other ways by other agencies.
- The Russian SID&GP noted issues under its inadequate stability program citation that there was an expectation for the company to “contact the Russian regulator,” which did not take place.
Each of the three regulators also shared their processes for how they evaluate and escalate inspection findings to determine if regulatory action is needed. Those processes are the subject of a follow-up article.
About The Author:
 Jerry is Govzilla’s senior GMP quality expert. He brings 40 years’ experience in the pharma industry, including 31 years at Eli Lilly, where he worked in product development, biosynthetic human insulin manufacturing, and site and corporate quality roles. In corporate quality, he designed and implemented a comprehensive GMP Intelligence process to identify, analyze, and archive pertinent drug GMP regulations, inspection findings, trends, and best practices in the U.S. and internationally. Chapman was founder and chairman of the GMP Intelligence subgroup of the Midwest Discussion Group from 2005 to 2010. He also served as senior editor at International Pharmaceutical Quality for six years; has been an invited speaker at PDA, AAPS, ISPE, and RAPS events; and was a consultant for the animal health and compounding pharmacy industries.
Jerry is Govzilla’s senior GMP quality expert. He brings 40 years’ experience in the pharma industry, including 31 years at Eli Lilly, where he worked in product development, biosynthetic human insulin manufacturing, and site and corporate quality roles. In corporate quality, he designed and implemented a comprehensive GMP Intelligence process to identify, analyze, and archive pertinent drug GMP regulations, inspection findings, trends, and best practices in the U.S. and internationally. Chapman was founder and chairman of the GMP Intelligence subgroup of the Midwest Discussion Group from 2005 to 2010. He also served as senior editor at International Pharmaceutical Quality for six years; has been an invited speaker at PDA, AAPS, ISPE, and RAPS events; and was a consultant for the animal health and compounding pharmacy industries.