
Commissioning & Qualifying: Facility Considerations For Tissue Engineering
By Paschal Iwuh, Ph.D., principal consultant, Pharmatech Associates—a USP company

Tissue engineering, also called regenerative medicine, focuses on curing chronic diseases. It is a broad area in medicine that incorporates research on self-healing where the body uses its own systems, sometimes with help from foreign biological material, to recreate cells and rebuild tissues and organs. Because this area of biotechnological innovation emphasizes cell manipulation, the equipment, systems, and material methods for a state-of-the-art tissue engineering facility are markedly different from a conventional biotechnology facility. To determine the costs and types of validation methodology needed for each facility, future operators or owners should understand the main differences.
This article compares the validation methodology (equipment and process validation) used for a conventional bulk drug substance biotech facility with a personalized medicine tissue engineering facility to explain how commissioning and qualification (C&Q) activities are conducted for each type and the validation costs attributable to each facility.
Tissue Engineering And Regenerative Medicine
Tissue engineering evolved from the field of biomaterials development and refers to the practice of combining scaffolds, cells, and biologically active molecules into functional tissues to assemble into constructs that restore, maintain, or improve damaged tissues or whole organs. Artificial skin and cartilage are examples of engineered tissues that have been approved by the FDA. Tissue engineering has led to the development of new methods for regenerative medicine to treat disease models.
The terms “tissue engineering” and “regenerative medicine” have become largely interchangeable, and the field continues to evolve. In addition to medical applications, some non-therapeutic applications use tissues as biosensors to detect biological or chemical threat agents and tissue chips that can be used to test the toxicity of an experimental medication.
By understanding how individual cells respond to signals, interact with their environment, and organize into tissues and organisms, researchers have been able to manipulate these processes to mend damaged tissues or create new ones.
The process begins with building a scaffold from a wide set of possible sources, from proteins to plastics. Once scaffolds are created, cells with or without a “cocktail” of growth factors can be introduced. If the environment is right, a tissue develops. In some cases, the cells, scaffolds, and growth factors are all mixed together at once, allowing the tissue to self-assemble.
Another method to create new tissue uses an existing scaffold. The cells of a donor organ are stripped, and the remaining collagen scaffold is used to grow new tissue. This process has been used to bioengineer heart, liver, lung, and kidney tissue. Such an approach holds great promise for using scaffolding from human tissue discarded during surgery and combining it with a patient’s own cells to make customized organs that would not be rejected by the immune system.
A Typical Tissue Engineering Facility
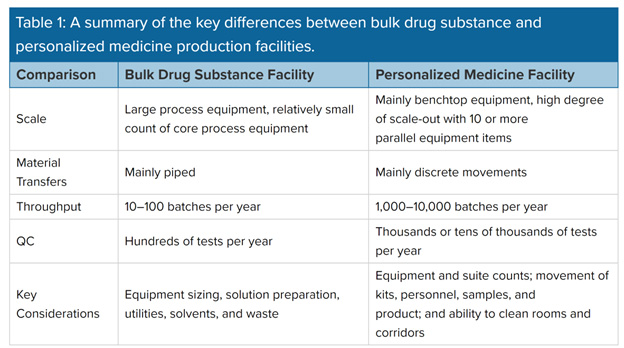
Tissue engineering and personalized medicine facilities produce thousands of small-scale batches per year. Inside the facility, researchers create and manufacture novel materials and devices for first-in-human use. Innovations from the facility should include implantable devices, tissue-engineered scaffolds, and tendon repair lubricants. Because their batches are smaller, with output measured in grams and emphasis on cell manipulation, the equipment, systems, and material methods differ from a conventional bulk drug or API-making facility. Thus, a different design approach is necessary for a facility geared toward material and personnel flows.
Various design factors should be considered for a personalized medicine facility to take care of 24/7 operations. The facility concept and operations are driven by a focus on product segregation, product traceability throughout the process steps, and an overall emphasis on product and patient safety.
One design driver concerns the ability of the facility to provide consistent high-quality therapies whenever the patient needs it. Such a facility would include features to minimize operational shutdown time for maintenance to offer 24/7 operations support (e.g., a robust HVAC concept, high-quality cleanroom wall/ceiling systems with low level of maintenance, etc.).
To streamline production, design models that focus on room and zone availability and movements can prevent bottlenecks in corridors, gowning areas, and airlocks.
A Typical Conventional Bulk Drug Facility
A conventional facility for bulk drug substances or small molecule API produces 20 or more batches per year and its design may have to accommodate many short campaigns to make intermediates, with any given reactor used for several different reactions. The complete process to make drug substance is generally broken up into many subprocesses, with the intermediates bulked and stored in between. There is normally a strong focus on requirements for product changeovers and line clearance, as well as on utilities, solvent storage, emissions, and waste.
With bulk biologics (e.g., mAbs and bacterial fermentation), the focus is on efficiency and throughput. Batches tend to be run straight through from vial thaw to drug substance formulation. Core equipment tends to be used once per batch. Design challenges revolve around the size and count of upstream trains and the sizing of downstream equipment, with a focus on balancing equipment size against utilization. The sizing and scheduling of solution preparation have to be taken into consideration.
Equipment/Systems Validation
Validation is defined by the FDA as establishing documented evidence that provides a high degree of assurance that a specific process or systems or equipment or procedure will consistently produce a product meeting its predetermined specifications and quality attributes. The keywords are “consistently” and “quality attributes” and there are four different types of equipment validation or qualification ranging from design qualification (DQ) to installation qualification (IQ), operational qualification (OQ), and performance qualification (PQ).
Systems used in tissue engineering facilities are smaller than those used in the bulk drug or API making facility, and they comprise freezers, incubators, refrigerators, and 10-L or 20-L reactors. These systems require only installation and operational qualification (IOQ) procedures to be successfully executed and conducted with approved final reports before being released for commercial purposes. Systems and equipment for bulk drug facilities are much larger and can include the following: lyophilizers, depyrogenators, fermenters, rinsers and washers, filtration devices, and 1,000-L to 2,000-L bioreactors. These systems require IOQ validation and design and performance qualification. This is an important difference between the two types of facilities.
Process Validation
By definition, process validation is the documented evidence that the process, operated within established parameters, can perform effectively and reproducibly to produce an intermediate or API that meets its predetermined specifications and quality attributes. The number of process validation runs for either the personalized or bulk drug substance facility depends on the complexity of the process or the magnitude of the process change being considered. Process validation should confirm that the impurity profile for each API is within the limits specified. The impurity profile should be comparable to or better than historical data and the profile determined during process development or for batches used for pivotal clinical and toxicological studies.
Bulk drug production requires large equipment with reactors in the thousands or tens of thousands of liters. Personalized medicine produces small-scale batches, typically no more than a few hundred grams, but it produces perhaps ten thousand such batches per year. As it is based on each patient's unique genetic makeup, personalized medicine is a more challenging process to validate because the cells or tissues are individually tailored to the patient.
For bulk drug process validation runs, prospective and concurrent validation are normally advised, with three consecutive successful production batches used as a guide, but there may be situations where additional process runs may be warranted to prove consistency of the process. Critical process parameters should be controlled and monitored during process validation studies. Process parameters unrelated to quality, such as variables controlled to minimize energy consumption or equipment use, need not be included in the process validation. Prospective validation should normally be performed for all API processes. Prospective validation performed on an API process should be completed before the commercial distribution of the final drug product manufactured from the API.
Validation Costs
The validation costs comprise the overall cost of commissioning and qualification (C&Q) of the facility and the validation/qualification of the equipment in each facility. Cost drivers for commissioning and qualification (C&Q) for a tissue engineering personalized medicine facility and a bulk drug facility include:
- Writing test protocols, test forms, and reports
- Confirming design input traceability
- Procuring parts for and performing QC testing on the test units
- Developing test software
- Designing and validating test fixtures
- Executing dry runs
- Analyzing data
Key Takeaways
The facility designs for tissue engineering and personalized medicine differ significantly from traditional bulk drug facilities. These innovative regenerative medicine facilities prioritize the flow of personnel and equipment, aiming to provide continuous operational therapy to clients around the clock. In contrast, conventional bulk drug facilities focus heavily on product changeover requirements, line clearance, utilities, solvent storage, emissions, and waste management.
Furthermore, validation of equipment and processes in these two types of facilities varies considerably due to their distinct operational needs. This divergence in validation approaches often leads to a favorable reduction in commissioning and qualification (C&Q) and validation costs for facilities dedicated to tissue engineering and regenerative medicine.
About The Author:
 Paschal Iwuh, Ph.D., is a principal consultant at Pharmatech Associates—a USP company. He has over 15 years’ experience in commissioning and qualification in the pharmaceutical, biotech, and food and beverage industries. He focuses on large-scale projects, with particular emphasis on new products, processes, and facilities. He recently led a team of commissioning agents and validation consultants through a major capital project (covering a 92,000 square foot facility) delivering impact and risk assessment, commissioning, qualification, and SOP generation for a CGT company transitioning from R&D to clinical production.
Paschal Iwuh, Ph.D., is a principal consultant at Pharmatech Associates—a USP company. He has over 15 years’ experience in commissioning and qualification in the pharmaceutical, biotech, and food and beverage industries. He focuses on large-scale projects, with particular emphasis on new products, processes, and facilities. He recently led a team of commissioning agents and validation consultants through a major capital project (covering a 92,000 square foot facility) delivering impact and risk assessment, commissioning, qualification, and SOP generation for a CGT company transitioning from R&D to clinical production.