
Cell Therapy: Process Design Considerations To Support Commercialization

By Herman Bozenhardt, Bozenhardt Consulting Services, and Erich Bozenhardt, IPS-Integrated Project Services
Cell therapies have the potential to revolutionize the biopharmaceutical world, but today’s processes, logistics, and delivery make for a challenging entry into the sector’s growth curve. As the industry evolves, we will have to answer (at least) three important questions when bringing these exciting new therapies to market:
- What benefit does the therapy provide in terms of patient longevity and product efficacy?
- What will the cost of the new therapy be, especially in the early stages of low volumes, and how can process manufacturing impact that cost?
- How can we, as an industry, provide the therapy that requires, in some cases, the patient to be resident/connected for cell collection and re-infusion, given that the scale of operation may force the providers to have only one national manufacturing site for the therapy?
Questions like this continue to propagate regarding the business, management, and ethics of an entirely new generation of healthcare, in the face of the evolving traditional market. Innovative and effective cell therapy treatments are advancing in the R&D pipeline, but cost and logistical challenges have been limiting the commercial success of the selected therapies that have been approved. The considerations on the road to commercialization for cell therapy take on a higher level of complexity than those for traditional biopharmaceuticals. These are the challenges that the manufacturing industry must face and provide solutions via logistical or biotechnology processes.
Defining Cell Therapy
The FDA’s Office of Cellular, Tissue and Gene Therapies (OCTGT), part of the Center for Biologics Evaluation and Research (CBER), is responsible for regulating cell therapy products, human gene therapy products, and some devices related to cell and gene therapy. (Though the line between what are devices and what are therapies is somewhat blurry.)
CBER recently introduced the Regenerative Medicine Advanced Therapy (RMAT) designation, intended to help reduce product development timelines for cell therapies and related products. In an FDA Voice blog post announcing the new designation, CBER director Peter Marks, Ph.D., explained its scope: “Sponsors of certain cell therapies, therapeutic tissue engineering products, human cell and tissue products, and certain combination products may obtain the RMAT designation for their drug product if the drug is intended to treat serious or life-threatening diseases or conditions and if there is preliminary clinical evidence indicating that the drug has the potential to address unmet medical needs for that disease or condition.” The program paves the way for accelerating the cell therapy pipeline and challenges the industry to move forward quickly and effectively with new therapies.
Regenerative medicine is not new — the industry has been producing human skin graft material for 20 years, by growing skin cells on a substrate. We have learned how to acquire cells, handle cells, anchor them, process the growth cycle, harvest them, and package and ship them. Genzyme was the first innovator this completely new field, and while its initial market was admittedly large, the task of establishing it was both daunting and complex. As we transition to the era of gene and cell therapies, we face additional questions and issues, even those as simple as distinguishing cell from gene therapy.
Referring back to the skin cell industry, what is the difference between a human skin graft derived from original skin cells and one derived from (human) stem cells? And how would you do a potency assay on an acellular tissue graft derived from stem cells? Modifying patients’ cells via adeno-associated virus in vivo is gene therapy, but using a different genetic engineering method ex vivo to produce a chimeric antigen receptor (CAR) T-cell is a cell therapy. The product and the claimed mechanism of action can drive these differentiations, even if the process is similar.
Autologous Therapies
Some companies are betting on autologous therapies as the quickest and safest route to bring new treatments to market. These therapies challenge us as an industry to deliver personalized medicine in a reliable and repeatable way. In the autologous therapy production lifecycle, the specific patient is both the raw material source and the consumer, so the focus is on the collection, processing, and re-infusion of cells for each patient. This forces us to design “operator-less” systems that can exist remotely and that unite single-use technologies and automated processes. Ready-to-use skids for scale-up and automation are critical to future successes.
Autologous Process Concepts
Some of the major concepts and solutions for developing the full lifecycle of autologous therapy production and administration are:
- Small contained and simple systems could utilize a fixed-bed culture technology with an existing scale-up/automation solution like the Nunc Cell Factory, Octane Biotech Cocoon, or Miltenyi Biotec CliniMACS Prodigy. The Nunc Cell Factory is a stacked 2D bioreactor system utilized in the vaccine industry, and Nunc offers a standard automated solution for handling commercial-scale quantities of 2D bioreactors. The Octane Cocoon and Miltenyi Prodigy are closed automated culture and purification systems for human cells.
- Developing the process to use closed systems can reduce the amount of clean room space required and mitigate the cross-contamination risk.
- Gaining a deeper understanding of cell culture needs — and determining how to meet those needs without a conventional incubator — can also aid in the commercialization of autologous therapies. Concerns about moisture loss through vent filters and CO2 control have restricted several processes to manual operations, since the cell line must be loaded into each incubator.
- Logistics and volume need to be considered during the development of the process for scale up and automation. Cell collection and processing is a key manipulation, and early technology development has yielded some new patient systems such as the Fenwal Amicus and Terumo Spectra Optia. These systems collect and process cell donations, so the desired fraction can be shipped frozen, with a significantly longer allowable storage time versus a refrigerated whole blood donation.
- As these (package-sized) donations arrive at the manufacturing facility, the management of what will ultimately become individualized lots needs to be established. Processing systems and documentation will likely need to mimic or utilize an individualized electronic batch record system (EBRS) to provide fidelity and assure quality with cellular materials. This system would likely be accessible at the collection point (local or centralized) and trace the material via the entire chain of custody (even FedEx shipment data acquisition- of time and temperature). At the end point, the EBRS will verify the route compliance and provide a “go” or “no-go” signal prior to re-infusion.
- Barcode readers have been standard in pharmaceutical supply chains for many years, and workarounds have been developed for frost-over issues in the cold chain; however, cryogenic-capable radio frequency identification (RFID) tags can provide a simpler solution for minimal system cost.
Get your process design questions answered in Erich and Herman Bozenhardt’s webinar:
Cell Therapy: Process Design Considerations To Support Commercialization
Allogeneic Therapies
While autologous therapies are more straightforward from a patient safety perspective, specifically addressing graft-versus-host disease, researchers have also developed allogeneic cell therapies — with common donor cells as the source of therapy for a spectrum of patients — that sufficiently address safety concerns. Allogeneic therapies relieve some of the logistical concerns, as a batch can provide therapy to many patients. However, donation logistics can be complex if the number of cell expansions is limited, requiring new donor material for every batch. Plus, the risk profile of each batch becomes a critical issue. In autologous therapies, the loss of one batch impacts one patient, but in the allogeneic manufacturing cycle such a loss impacts many intended patients. Thus, the need for automation, tracking, and reduced human intervention becomes the key process design criteria.
Allogeneic Process Concepts
From a process viewpoint, induced pluripotent stem cells (iPSCs) have the potential to support a traditional cell-banking approach to human stem cell based therapies. Some allogeneic therapies take a different approach, instead utilizing bacteria cell for the therapy.
One common commercialization consideration for any type of allogeneic therapies is development of the culture to use microcarriers. Culturing on suspended microcarriers instead of 2D bioreactors significantly reduces the cost of goods and capital cost of the facility, enabling larger-scale production due to reduce facility footprint requirements. Lonza has done extensive process development in this area and has designed a technology platform that can be applied to a variety of cell therapies. The platform is based on mesenchymal stem cell (MSC) lines attached to micro-carriers, where media is perfused during culture and either a tangential flow filter or kSep centrifuge is used to remove and wash the cells. Others are exploring this type of platform but have not published their findings extensively.
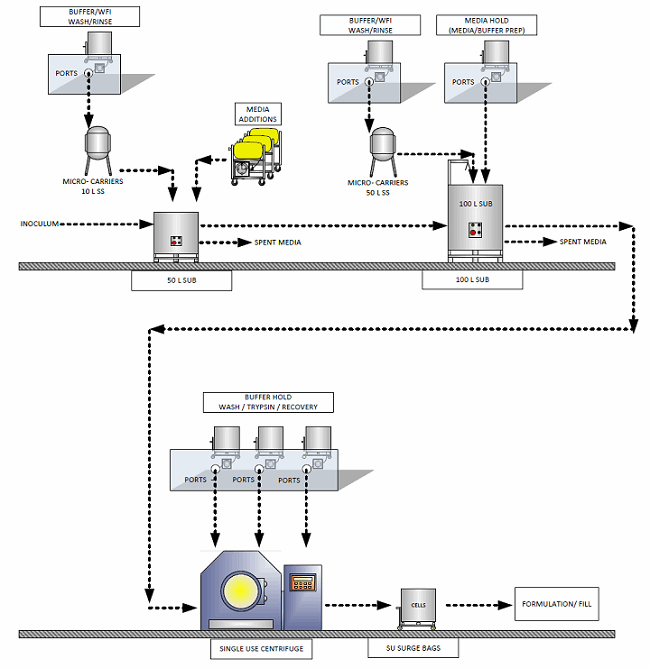
Figure 1: Flow diagram of a microcarrier process
Some cell therapies achieve efficacy through cell secretions (e.g., cytokines, exosomes) rather than through the cell itself. These factors have the potential to be easier to manufacture and less demanding to store than whole live cells. Perfusion through a large (non-benchtop) 3D fixed bed bioreactor — like Pall’s iCELLis — or through a stirred bioreactor with microcarriers can enable a compact, large-scale commercial system.
As with autologous therapy, cold chain logistics for allogeneic therapy shifts away from the traditional model of refrigerated cases of finished product being handled by a distributor, to shipping individual cryogenic units of sale. Other new considerations could include administration or infusion through a medical device or a preconditioning treatment, which are as much part of the process as operations in an environmentally controlled manufacturing facility. The basic concepts of EU GMP Annex 1 and Annex 2 will serve us well in the design of these facilities, along with ensuring our processes work within the production environment. In the case of allogeneic therapy, the operating costs will be minimal compared to the extraordinary value of the therapy to the patient.
Other Optimization Opportunities
All cellular therapies share certain common process considerations to ensure a repeatable and robust process. Media recirculation improves efficiency and quality (batch uniformity), and in combination with on-line monitoring, can optimize media use. We expect to see a functional reduction of serum in cell therapy processes, which will not only reduce process costs, but more importantly will reduce process/product variability and requirements of viral clearance and contamination.
Cell therapies have the potential to open new avenues of treatment for numerous critical diseases. Our industry needs to position these processes/products/systems for long-term success by reducing the cost to patients, improving availability of these therapies to patients, assuring the quality of product delivery, and developing practical logistical solutions. Holistically developing processes for scale-up and automation across manufacturing and administration will drive down costs and improve logistics for cell therapies.
For further discussion of facility-specific considerations for cell therapy manufacture, read the article Designing The Cell Therapy Facility Of The Future.
About The Authors:
 Herman Bozenhardt has 40 years of experience in pharmaceutical, biotechnology, and medical device manufacturing, engineering, and compliance. He is a recognized expert in the area of aseptic filling facilities and systems and has extensive experience in the manufacture of therapeutic biologicals and vaccines. His current consulting work focuses on the areas of aseptic systems, biological manufacturing, and automation/computer systems. He has a B.S. in chemical engineering and an M.S. in system engineering, both from the Polytechnic Institute of Brooklyn.
Herman Bozenhardt has 40 years of experience in pharmaceutical, biotechnology, and medical device manufacturing, engineering, and compliance. He is a recognized expert in the area of aseptic filling facilities and systems and has extensive experience in the manufacture of therapeutic biologicals and vaccines. His current consulting work focuses on the areas of aseptic systems, biological manufacturing, and automation/computer systems. He has a B.S. in chemical engineering and an M.S. in system engineering, both from the Polytechnic Institute of Brooklyn.
 Erich Bozenhardt is the process manager for IPS-Integrated Project Services’ process group in Raleigh, NC. He has 10 years of experience in the biotechnology and aseptic processing business and has led several biological manufacturing projects, including cell therapies, mammalian cell culture, and novel delivery systems. He has a B.S. in chemical engineering and an MBA, both from the University of Delaware.
Erich Bozenhardt is the process manager for IPS-Integrated Project Services’ process group in Raleigh, NC. He has 10 years of experience in the biotechnology and aseptic processing business and has led several biological manufacturing projects, including cell therapies, mammalian cell culture, and novel delivery systems. He has a B.S. in chemical engineering and an MBA, both from the University of Delaware.
Photo of the iCELLis bioreactor supplied courtesy of Pall Life Sciences