
Antibody Drug Conjugates (ADCs): Complex, Powerful Drugs Requiring Complex Analytical Solutions
: Complex, Powerful Drugs Requiring Complex Analytical Solutions")
By Mario DiPaola1 and Andrew Hanneman2
The concept of targeting cytotoxic drugs to selectively destroy tumors is not new. Indeed, as early as 1913, Paul Ehrlich proposed the concept of a “magic bullet” for delivering cytotoxic substances to tumor tissue by means of targeting agents. Approximately 50 years later, Mathe, G. et al. (1958) linked methotrexate to a polyclonal antibody targeting leukemia cells. In subsequent years, this approach was further evolved by replacing polyclonal antibodies with mouse monoclonal antibodies (mAbs) produced by hybridoma technology and then eventually with humanized mAbs. Unfortunately, these early ADCs displayed limited clinical success due primarily to immunogenicity issues, low drug potency, toxicity, and instability.
In the past decade, much focus has been placed on developing more stable linkers and more potent cytotoxic payloads; these improved constructs have resulted in better therapeutic agents, as with the recent approvals of Adcetris, an anti-CD30 auristatin conjugate, and Kadcyla, an anti-Her2 maytansine conjugate. In spite of these improvements, further optimization of linkage sites on the antibody, along with higher potency cytotoxins, better antigen selectivity, and improved cleavable and non-cleavable linkers have resulted in better, highly-targeted oncology drugs.
The new generation of ADCs aims to maintain the drug to antibody ratio (DAR) to be as homogeneous as possible; also, rather than linking non-specifically to functional groups on the antibody, the linkages are site specific through engineered cysteines (THIOMAbs) or C-terminal selenocysteines, aldehyde tags introduced by formylglycine generating enzyme (FGE), as well as the incorporation of unnatural amino acids, such as p-azido phenylalanine, p-acetyl phenylalanine, or propynyl tyrosine.
Currently, there are nearly 50 different ADCs in various stages of clinical development spanning from Phase 1 up to Phase 3. A number of these drugs are being tested for treatment of hematological malignancies, with focus mostly on acute and chronic myelogenous leukemia, followed by non-Hodgkin lymphoma. There are also a number of ADCs under evaluation for the treatment of solid tumors, especially breast cancer.
The design and development of complex molecules, such as ADCs, require sophisticated analytical methodologies to allow for complete characterization of these molecules. These methodologies are necessary to determine the ratio of drug to antibody. The site of drug linkage can also be identified and the relative level of occupancy at each linkage site can be measured. Methods need to be sensitive enough to detect and measure any free drug (warhead), as well as free linker or related non-product components, and be stability-indicating to assess the long-term stability of an ADC.
Schematically the extent of testing involved for the production and release of an ADC product is summarized in Figure 1.
Figure 1: Analysis of ADC product and its constituents
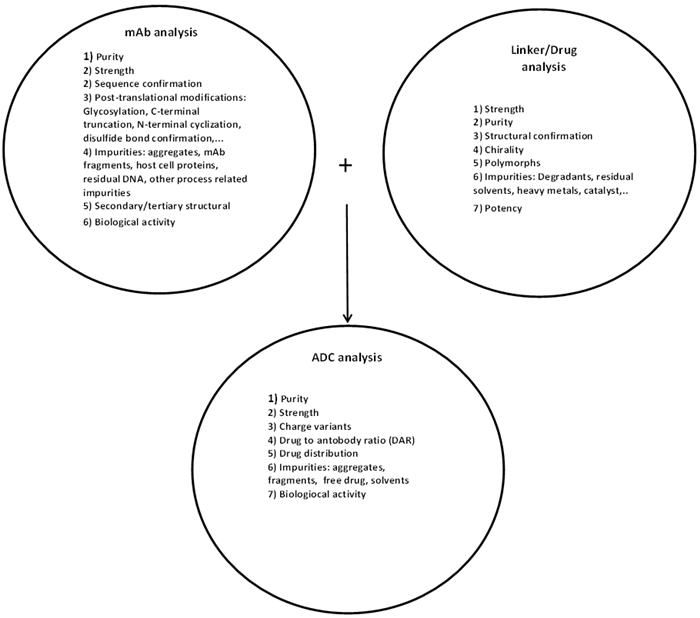
As summarized in Figure 1, prior to manufacturing an ADC, extensive testing must be performed on the constituent antibody along with the linker and the cytotoxic warhead. These tests include assessment of identity, purity, and impurities (process and product-related), strength, biological activity, safety with respect to bioburden and endotoxin, as well as stability.
One of the more important attributes to test for an ADC product is the drug-to-antibody ratio, as this affects drug clearance, safety, and, of course, potency (efficacy). A DAR that is too high will potentially cause toxicity and possibly faster clearance. A DAR that is too low will result in limited efficacy. Due to their high molecular weight, obtaining a DAR value for an intact ADC requires the use of a high resolution mass spectrometer (MS), such as a Time of Flight (ToF), or hybrid-ToF instrument. While electrospray ionization (ESI) MS coupled with HPLC will yield very accurate mass determinations, there is an inherent assumption in using this approach in that the relative MS ionization efficiencies are equivalent for each DAR species present. As such, DAR quantification may be more accurate if the various DAR species are chromatographically separated prior to mass measurement, wherein UV detection may be additionally employed to obtain the DAR determination. Achieving chromatographic separation at the intact, or “top-down” mass analysis level, however, may be challenging. An additional challenge for intact mass analysis is the preservation of any noncovalent interactions, if present. In such a case, it is necessary to perform LC-MS under native pH ESI conditions to preserve these less stable interactions.
Accurate quantification of the payload stoichiometry by MS may be more practical, yet relatively simple, by employing a “middle-down” LC-MS approach. A facile reduction step, yielding light chain (LC) and heavy chain (HC) separately (“2-part” middle down), has been shown in many cases to allow for chromatographic separation of LC and HC and their various conjugates (i.e. LC+1, LC+2, HC+1, HC+2, etc.). By including a single enzymatic step, IdeS, prior to reduction, an additional cleavage of HC, into Fc and Fd, yields a “3-part” middle down LC-MS approach for DAR determination.
Another equally important ADC attribute is the DAR distribution which provides a measure of the homogeneity of drug linkage sites across the molecule. To map linkage sites and establish the relative site occupancy, it is necessary to use a “bottom-up” or peptide mapping methodology. For this, the ADC is first deglycosylated, then, depending on linkage chemistry, reduced and alkylated and enzymatically or chemically (however, less preferred) digested, prior to separation by RP-HPLC, typically using a C18 column, and analysis by ESI-MS/MS. Utilizing such an approach will allow for identification of linkage sites and homogeneity of linkage across the sites.
Of course, peptide mapping will also reveal other post translational modifications including deamidation, oxidation, N-terminal cyclization through pyroglutamic acid formation, isomerization, glycation, C-terminal truncation, and disulfide shuffling. Localization of actual drug conjugation sites and any PTMs is further enhanced by MS/MS fragmentation approaches including high and low energy collision-induced dissociation (CID) and Electron Transfer Dissociation (ETD). The combined use of top-down, middle-down, and bottom-up MS approaches can yield a comprehensive and relatively complete ADC characterization package.
More recently, ion mobility-mass spectrometry (IM-MS) has been gaining traction for its use in the analysis of ADC molecules. Ion mobility provides an additional dimension of separation, as it allows for separation of gas-phase ions based not only on size, but also the molecular shape or cross sectional area. With ion mobility complementing mass measurement, the combination can provide substantially improved resolution, yielding a plethora of structural information (Lanucara et al., 2014) for complex molecules, such as ADCs.
While high resolution ESI-MS can yield quick and accurate DAR values and profiles, it may be possible to use alternate, simpler analytical techniques to obtain such DAR values. For drugs with unique UV/Vis spectra, it is possible to determine the DAR by simply applying UV/Vis spectroscopy to measure both the drug-specific absorbance and the antibody absorbance at 280 nm and then converting such absorbances to concentrations using the respective extinction coefficients. Furthermore, for many ADC constructs, the use of hydrophobic interaction chromatography (HIC) can allow for resolution of the different ADC species, each carrying varying payload stoichiometries (Hamblett et al., 2004).
As with most biopharmaceutical products, aggregation of ADCs is a significant concern and must be properly characterized and measured. Frequently, SEC is the method of choice. However, this method is potentially prone to inaccuracies due to aggregate loss by adsorption (Stulik, K., et al., 2003) or outright filtration of any aggregated species exceeding the SEC resin pore size. It is, therefore, recommended that alternate techniques such as analytical ultracentrifugation (AUC) or field-flow fractionation (FFF) be used to confirm SEC results.
As part of the impurity analysis for an ADC, it is critical to have specific and sensitive methods that allow for detection, characterization, and quantification of any free cytotoxic warhead or linker-related species. Often these impurities can be simply separated and detected by RP-HPLC with UV detection; however for greater sensitivity and specificity, use of LC-MS (QQQ) is preferable to UV detection alone.
The conjugation of cytotoxic drugs to antibodies will increase the hydrophobicity of the antibody; this change can be destabilizing and lead to lower product solubility and shorter product shelf-life. To assess the effects of conjugation, there are a number of biophysical techniques available for use. Application of circular dichroism (CD) for analysis of an ADC relative to the unconjugated antibody can provide information on changes related to secondary structure (far UV) and tertiary organization (near UV). Similarly, differential scanning calorimetry can be used to determine the transition temperatures at which various structural domains within the protein undergo unfolding.
A more comprehensive summary of how different analytical methodologies may be used to evaluate the various product attributes of ADCs is provided in Table 1.
Table 1: Summary of analytical methods and their application for analysis of ADCs
|
Product attributes |
Analytical methodologies |
|
General and Physico-chemical |
|
|
Strength |
|
|
Functional/Potency |
|
|
Identity |
|
|
Purity and Impurities |
|
|
Secondary/tertiary structure |
|
As tabulated in Table 1, there are many analytical methods available to thoroughly analyze complex molecules such as ADCs. The application of these methods can yield the relevant product information needed by ADC developers to improve manufacturing processes, formulations, and to identify a subset of methods that can be used for product release and stability assessment.
References:
Mathe, G. et al. (1958) Effect sur la leucemie L 1210 de la souris d’une combinaison par diazotation d’A-methopterine et de gamma globulines de hamsters porteur de cette leucemie par heterogreffe. C. R. Acad. Sci. 246, 1626-1628.
Hamblett, KJ, Senter, PD, Chace, DF, Sun, MM, Lenox, J, Cerveney, CG, et al. (2004) Effects of drug loading on the antitumor activity of a monoclonal antibody drug conjugate. Clin. Cancer Res. 10, 7063-7070.
Lanucara, F, Holman, WS, Gray, JC, Eyers, EC (2014) The power of ion mobility-mass spectrometry for structural characterization and the study of conformational dynamics. Nature Chemistry 6, 281-294.
Stulik, K, Pacakova, V, Tich, M (2003) Some potentialities and drawbacks of contemporary size exclusion chromatography. J. Biochem. Biophys. Methods 56(3), 1-13.
Authors:
1: Mario DiPaola*, Chief Scientific Officer, Blue Stream Laboratories, Inc. Woburn MA 01801
2: Andrew Hanneman, Associate Directory of Mass Spectrometry, Blue Stream Laboratories, Inc. Woburn MA 01801
*E-mail address: mdipaola@bluestreamlabs.com and to whom correspondence should be addressed