
An Introduction To Forced Degradation Studies For Drug Substance & Drug Product
By Vinubhai N. Patolia

The forced degradation study is considered a vital analytical aspect of the drug development program for small molecules. Forced degradation, commonly known as stress testing, is carried out to demonstrate as specificity to developed a stability-indicating analytical method, using high-performance liquid chromatography (HPLC), i.e., a single analytic method that is capable of separating the degradant peaks from the drug substance/drug product peak. As per International Conference on Harmonization (ICH) guidelines (Q1A), stability studies need to be performed to propose the shelf life of new drug substances and/or drug products. Shelf life studies are part of various regulatory submissions to the FDA.
Generally, three kinds of stability studies need to be performed in order to propose the shelf life of a drug substance and/or drug product: accelerated stability (ACC), intermediate stability (INS), and controlled room temperature (CRT) stability. The duration of accelerated study is about six months, intermediate stability and controlled room temperature stability studies take about 12 to 24 months. A stability study is conducted to determine the intrinsic stability of the molecule and, during the study, it is expected the drug substance/drug product will degrade/decompose and generate other molecules, which are known as impurities. During the forced degradation studies, various stress conditions are deliberately applied to degrade/decompose the main compound and generate impurities, which should separate from the main compound and from each other. Therefore, forced degradation studies are a tool to estimate the degradant/decomposed impurities that would occur during stability studies and are used to propose the shelf life of the new drug substances and/or drug products.
Various analytical techniques/equipment can be used to separate and estimate all the degradant compounds expected to be present at the time of the forced degradation study. HPLC with UV detector (HPLC-UV) and photodiode array detector (HPLC-PDA) are well-known techniques and are widely used in pharmaceutical industries during forced degradation studies as part of the development and validation of the stability-indicating method. HPLC with mass detector (LC-MS), gas chromatography with mass detector (GC-MS), and nuclear magnetic resonance (NMR) spectroscopy are important techniques to identify the structure of the degradants.
Important Applications Of The Forced Degradation Study
Forced degradation is a critical analytical study for the development of stability-indicating methods to be used by pharmaceutical companies as part of regulatory submissions to the FDA. Some of the applications of the studies are:
- To develop and validate stability-indicating methods as per ICH guidelines.
- To identify structure and toxicity and to set up specification of degradants or impurities.
- To propose shelf life of the product without real-time stability information.
- To optimize formulations and to select placebos for drug product to avoid interference.
- To justify impurities that are process related or degradation products.
- To support identification of root cause during out-of-specification (OOS)/lab investigations.
- To accompany drug master file and ANDA/NDA and IND submissions to the FDA.
Selection And Procedures Of Forced Degradation Condition
As per ICH guidelines and common industry practice, forced degradation is usually performed in different stress conditions, i.e., acid, alkali, peroxide, thermal, and UV, along with a control sample. There are no industrial guidelines about how much degradation should be achieved; however, per current industrial practices, 5 to 30 percent degradation should be achieved in any one of the applied stress conditions. The aim of the degradation to be achieved through stress testing is to mimic the control room temperature stability conditions. In cases where higher or lower degradations are observed, the conditions or concentrations of the reagent should be optimized. Mass balance should be demonstrated during the degradation study and it should be around 100 percent, taking into consideration margins of analytical errors. All the degradants/impurities must be calculated during mass balance evaluations.
During the forced degradation study, any batch that will not be the part of regulatory submission can be used. In the case of a drug product, if multiple strengths are available with the same placebos and different amounts, the strength that has highest ratio of placebo vs. active pharmaceutical ingredient (API) should be used. Where placebos are different, then forced degradation of all the strengths must be demonstrated. During the drug product degradation study, both the placebo and API must be demonstrated to identify actual degradation pathways. Where placebos are different for different strengths of drug product, then all the placebos should be considered for the degradation study.
The following (Table 1) are the suggested degradation conditions as per current industrial practices and accepted by the FDA during DMF/ANDA/NDA and IND submissions for regulatory approval.
Table 1: Suggested Experimental Conditions For Forced Degradation Studies
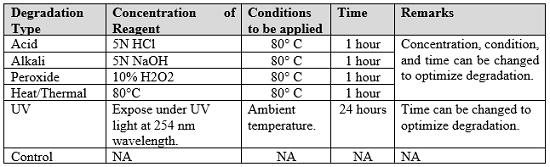
Degradation should be performed in solid or solution form, but it is recommended it be performed in solution form using the diluent/mobile phase to obtain a homogeneous effect. Degradation studies can be initiated with harsh conditions (i.e., high concentration of reagent with high temperature) to shorten the time of the study. In cases where degradation of more than 30 percent is found, milder conditions should be applied by reducing the concentration of the reagent, lowering the temperature, etc. Degradation conditions shall be optimized to achieve a target based on the initial degradation outcome. pH should be adjusted to about 7.0 for acid and alkali degradation to extend the shelf life of the chromatographic column. If degradation is not found in any of the above conditions, different reagents and conditions can be applied, e.g., H2SO4, Zn, etc. There are a few molecules that do not degrade under any harsh conditions and they are therefore considered rock stable molecules. This kind of molecule will not generate any additional impurities/degradant peaks during a stability study.
Characterizations And Mass Balance Of The Forced Degradation Study
All the solutions should be analyzed using pre-developed suitable analytical methods, along with the diluent, placebo, and control sample solutions. The suggested acceptance criteria during the forced degradation should be as follows:
- Degradation of at least 5 to 30 percent should be observed in any of the conditions.
- The main peak must be well separated from the diluent, placebo, known peaks, and degradant peaks generated during degradation.
- The main peak must be pure, i.e., no other peak should be merged with the main peak. Generally, it can be determined using software such as Empower 3 if the purity threshold is greater than the purity angle, which means the peak is pure. Advanced software can generate a 3D picture of all the peaks from each chromatogram, which helps to identify peak purity, as shown in following picture.

Note: It is possible that the main peak is separated from all the peaks and is pure; however, known impurities or degradant peaks generated during the degradation study are not pure and may be merged with other peaks. In that case, attempts should be made to modify the analytical method to separate all the peaks from each other. Therefore, it is also recommended that peak purity should be evaluated for all peaks (main peak, known impurity peaks, and all the degradant peaks) to avoid consequences during the stability study. When attempts to obtain peak purity of the degradant peaks fail, the method development details should be documented and a risk-based approach should be applied to accept the method as stability-indicating.
During the forced degradation, many more degradant peaks can be generated from the main (active) peak. The percent degradation of the main (active) peak shall be calculated after completion of the analysis. The following formula is generally used in pharmaceutical industries and accepted by the FDA to calculate the percent degradation.
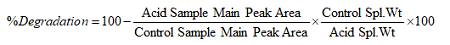
Using the percent degradation, mass balance should be evaluated for all the degradant peaks. The mass balance should be about 100 percent, considering margin of errors during analysis.
HPLC with mass detector (LC-MS and LC-MS/MS), gas chromatography with mass detector (GC-MS), and nuclear magnetic resonance (NMR) spectroscopy are important techniques to identify the structure and mass balance of the degradants. The characterization of the degradants can be determined using advanced analytical techniques such as LC-NMR. Thin layer chromatography (TLC), column chromatography, and preparative HPLC can be used to isolate the impurities from residue or filtrate generated during the API manufacturing process.
Development Of Stability-Indicating Methods Incorporating Force Degradation Study
During the forced degradation study, degradation products can be generated that might be more than expected during the ACC. Degraded samples, along with known impurities, must be used to develop the analytical method. These samples will represent the worst-case scenario and all the peaks (main peak, degraded peaks, and known impurities) must be well separated from each other. In cases where the analytical method is not able to separate all the peaks, the method needs to be redeveloped to achieve separation of all the peaks. Reference to books, literature, and experience is a key factor in developing the analytical method. One of the reputable books is Practical HPLC Method Development, by R. Snyder. It will help users to understand how to get better separation of all the peaks. Once an analytical method is developed that is capable of separating all the peaks from degraded samples, it means this method is capable of analyzing all the stability samples and, therefore, this analytical method will be considered stability-indicating. Method validation must be performed to demonstrate that the analytical method is suitable for its intended purpose (refer to ICH guidelines Q2 (R1) Validation of Analytical Procedures). The forced degradation information can also help with determining the root cause during OOS/lab investigations during the sample analysis, e.g., if any unexpected peaks are observed during the sample analysis that were not observed during any forced degradation condition, it can be concluded that the peak is due to contamination, either during the manufacturing process or during the analysis.
Conclusion
Forced degradation is an essential study that provides the knowledge and judgment to develop a stability-indicating analytical method. This study must be performed, and it should be part of regulatory submissions. This study also helps to establish the specification and shelf life of a drug substance or drug product. The information derived from the study will help to improve the formulation, manufacturing process, and storage conditions of the product. In addition, the forced degradation study also helps to discover root causes or any potential contamination during the manufacturing process or during laboratory analysis. Therefore, the forced degradation study must be demonstrated at the time of method development and before submission of the regulatory dossier to the FDA.
References:
- ICH Q1A (R2) Stability Testing of New Drug Substances and Products https://www.ema.europa.eu/en/documents/scientific-guideline/ich-q-1-r2-stability-testing-new-drug-substances-products-step-5_en.pdf
- ICH Q2 (R1) Validation of Analytical Procedures https://www.ema.europa.eu/en/documents/scientific-guideline/ich-q-2-r1-validation-analytical-procedures-text-methodology-step-5_en.pdf
- ICH Q3A (R2) Impurities in New Drug Substances https://www.ema.europa.eu/en/documents/scientific-guideline/ich-q-3-r2-impurities-new-drug-substances-step-5_en.pdf
- ICH Q3B (R2) Impurities in New Drug Products https://www.ema.europa.eu/en/documents/scientific-guideline/ich-q-3-b-r2-impurities-new-drug-products-step-5_en.pdf
- ICH Q6A Specifications: Test Procedures and Acceptance Criteria for New Drug Substances and New Drug Products (Chemical Substances) https://www.ema.europa.eu/en/documents/scientific-guideline/ich-q-6-test-procedures-acceptance-criteria-new-drug-substances-new-drug-products-chemical_en.pdf
- USP General Chapter <1225>: Validation of Compendial Procedures
- Empower PDA Software Manual — Getting Started guide
- Snyder, L., Kirkland, J., and Glajch, J. Practical HPLC Method Development. Wiley, 1997.
- Beginners Guide to Liquid Chromatography (Waters Series), 1st Edition. Waters Corporation, 2014.
- Ahuja, S., and Rasmussen, H. HPLC Method Development for Pharmaceuticals. Academic Press, 2007.
- Kats, R. “Forced Degradation Studies: Regulatory Considerations and Implementation.” BioPharm International, Jul. 01, 2005.
- Reynolds D., et al. “Available Guidance and Best Practices for Conducting Forced Degradation Studies.” Pharmaceutical Technology, Feb. 1, 2002.
About The Author:
 Vinubhai Patolia has 20 years of experience in analytical R&D and quality control in the pharmaceutical industry. After completing his M.S. degree (organic chemistry), he worked at Zydus Cadila, Sun Pharmaceutical Advanced Research Center, and Actavis Pharma. Most recently, he was manager of analytical R&D for Sun Pharma in Cranbury, NJ. During his career, Patolia has gained significant experience in method development, method validation, method transfer, and responding to FDA deficiencies for drug substance, drug product, and excipients for ANDA/NDA submissions. He has worked across various dosage forms, including solid oral dose, topical, suspension, and injectable.
Vinubhai Patolia has 20 years of experience in analytical R&D and quality control in the pharmaceutical industry. After completing his M.S. degree (organic chemistry), he worked at Zydus Cadila, Sun Pharmaceutical Advanced Research Center, and Actavis Pharma. Most recently, he was manager of analytical R&D for Sun Pharma in Cranbury, NJ. During his career, Patolia has gained significant experience in method development, method validation, method transfer, and responding to FDA deficiencies for drug substance, drug product, and excipients for ANDA/NDA submissions. He has worked across various dosage forms, including solid oral dose, topical, suspension, and injectable.