
Adoption Of Single-Use For Final Filtration
By Terri Love and Youness Cherradi

Introduction
Single-use systems (SUS) are now used routinely in the manufacture of biologics in both upstream and downstream operations. The biopharmaceutical industry has seen increased use of these technologies over the past decade and this trend continues with a global market expected to grow to over $4.3 billion by 2021.1
A recent development is the adoption of single-use assemblies for final filtration. These assemblies have evolved from the first simple assemblies of a capsule with tubing to more complex designs, which enable pre-use integrity testing, blow-down and sampling. Among others, GSK (Barnard Castle, U.K.)2 and Roche Diagnostics GmbH (Mannheim, Germany)3 successfully implemented single-use assemblies in their final fill process steps.
At GSK, the main driver for moving from stainless steel to SUS was the need to rapidly meet the intense demands of influenza vaccine production. SUS offered flexibility, robustness, and speed of implementation, and while cost reduction was not the primary concern, SUS were operationally efficient by minimizing the clean-in-place and sterilization needs associated with a multiuse product facility. Implementation of SUS at GSK reduced the campaign fill time from 36 to 12 hours, which led to a 40% improvement in capacity, allowing GSK to manufacture more batches and meet future demand for vaccines.2
Filtration strategy
Any filtration strategy requires consideration of the goal in the context of the specific feed stream. In the case of final filtration, the goal is to remove particles, including micro-organisms, from a purified and formulated drug product without changing product quality attributes and with minimal product losses.4-6 SUS for final fill are designed to simplify integration of sterilizing-grade filters into a fill line enabling robust, efficient sterilizing filtration of product.
Manufacturing controls throughout the process should minimize bioburden in the product and deliver it to the filling line with quality attributes within a consistent range. Bioburden increases the microbial challenge to the sterilizing-grade filter and contributes impurities which could lead to degradation of the drug product. The specifics of the upstream and downstream operations will determine whether a bioburden reducing filter is required before sterilizing filtration.
A bioburden limit should be established prior to sterilizing filtration.4 According to the EMEA-CPMP Note for Guidance on Manufacture of the Finished Dosage form, ‘In most situations not more than 10 colony forming units (CFU)/100 mL will be acceptable, depending on the volume to be filtered in relation to the diameter of the filter. If this requirement is not met, it is necessary to use pre-filtration through a bacteria-retaining filter to obtain a sufficiently low bioburden’. 7 Generally a bioburden reducing filter does not have to be validated to the absolute standards of the final sterilizing-grade filter, but it should be validated for robust performance as any other process step.
The design of the filtration train is further determined by the possible need for redundant filtration. A redundant filtration train is one in which there are two sterilizing-grade filters in series; the filter closest to the fill line is the primary filter and the second filter acts as a back-up to the primary filter in the event of a post-use integrity test failure. In a SUS, this configuration is generally referred to as single-use redundant filtration (SURF). The FDA recommends that ‘the use of redundant sterilizing filters should be considered in many cases’.4 In Europe, the guidelines for final filtration state that 'a second sterilizing-grade filter immediately prior to filling may be advisable'.6 Both filters should be subjected to pre-use, post sterilization integrity testing (PUPSIT) and both filters should pass this integrity test.
Post-use, if the primary filter passes the integrity test, there is no requirement to test the second filter. However, if the primary filter fails this integrity test, the second filter should be tested and, if this filter passes, the batch may be released.
The choice between using a bioburden reducing filter, a single sterilizing filter, or redundant sterilizing filtration depends on the specific process, value of the product, and history of filter test failures on site. Risk assessments help determine which filter train is the best option for each process.
While the membrane in each filter determines the retention capabilities, the design of the filter capsule should also be considered as this affects both the risk of contamination and also product losses due to holdup volume. Smaller filters inherently have lower holdup volumes, but capsule design plays a key role. For a given filtration area, stacked disk filters can reduce product losses by up to a factor of five compared to more traditional pleated filters; an important consideration when working with high value products.
Pre-use integrity testing and maintaining sterility
If the final filtration system fails to perform efficiently, there is a risk of leakage and contamination of the pharmaceutical drug product and redundant filtration can mitigate this risk. Pre-use integrity tests are particularly important since they confirm all components are working properly before use, however they can prove challenging to implement. In pre-use testing, the downstream side of the filter must remain sterile during the wetting and testing steps.5 This becomes more demanding when the two filters in a redundant assembly must be tested and the connection between the two sterilizing-grade filters must maintain sterility. There are several options for forming a sterile boundary downstream of the filter each with its own merits and degree of complexity (Table 1).
Table 1: Sterile boundary options
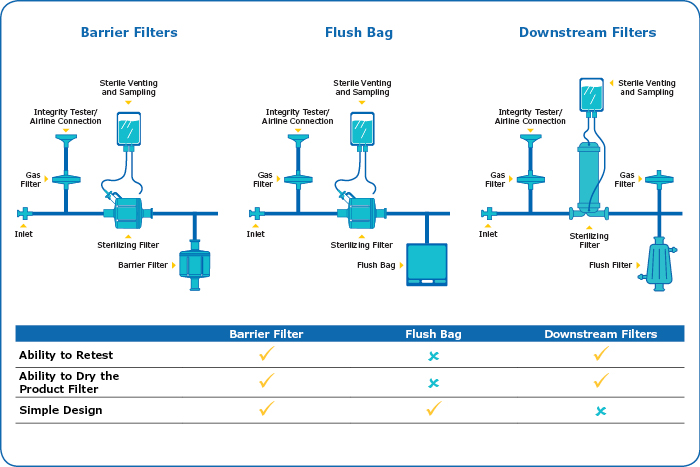
In general, the design of the sterile boundary will depend on whether a pre-use integrity test is performed, whether a blow-down of the filters is required, and the size of the sterilizing-grade filter.8 When no blow-down is required the simplest sterile boundary is a flush bag. However, when running a pre-use test with this arrangement, the number of re-tests is limited by the size of the flush bag so the bag should be sized to allow multiple flushes and re-testing. Depending on the size of the filter and number of retests, at some point this will result in a large and impractical bag size. 7,11 Generally, flush bags are useful for small area filters but for larger area filters, a downstream filter arrangement will work better.
There are two options for a downstream filter arrangement. The first is barrier filters which contains both hydrophobic and hydrophilic membranes. These sterilizing-grade filters allow an unlimited volume of water to be flushed through the hydrophilic membrane, while the hydrophobic membrane allow the pre-use integrity test to be performed without compromising sterility of the downstream side of the filter arrangement. This filter also enables blow-down of the sterilizing-grade filter. 9,10
For sterilizing-grade product filters of 20-inch size and above, it is recommended to use the air diffusion test rather than a bubble point integrity test if using barrier filters. The higher water pressures needed for proper wetting and integrity testing of these larger sterilizing-grade filters can result in the hydrophobic membrane layers becoming wetted out and can lead to false integrity test failures. Air diffusion use lower pressures, which will work with a simple wetting procedure and are more suitable for barrier filters.11
If it is necessary to run a bubble point test on the large area sterilizing-grade filter, then a downstream arrangement of a single hydrophobic gas or vent filter and a single hydrophilic filter can be used (Table 1). This assembly is more complicated than the other options, particularly if it is a SURF assembly, as two sets of hydrophobic vent and hydrophilic filter arrangements would need to be built in.
SURF Assembly Design
The filtration process requires decisions on the number and types of filters, whether a pre-use test is required and what kind of sterile boundary is necessary for the assembly. An example of a redundant assembly is shown in Figure 1. In this assembly, there are flush bags connected to the sterilizing filter vents to contain liquid during the venting process. A gas filter is connected before each sterilizing filter, enabling an integrity tester to be attached, while maintaining a sterile connection during the integrity testing of each sterilizing filter. Barrier filters allow the user to wet the filters and enable integrity testing and drying of each sterilizing filter.
After integrity testing, the filtration assembly will be flooded with water and, to prevent dilution of the product, the filter assembly should be blown down to remove residual water. During this operation it is important to exceed the bubble point pressure of the sterilizing filter by 0.5 – 1 bar, to displace all the liquid in the pores of the filter. For more efficient drying, the assembly can be designed to allow sequential drying of the sterilizing filters (Figure 1). Before drying, components should be assessed to ensure they can handle the applied pressures during flushing, integrity testing, blow-down and filtration. During blow down, care should be taken not to exceed the maximum allowable pressure of the “weakest” link in the assembly.8,10,12,13
Figure 1: Single use redundant assembly
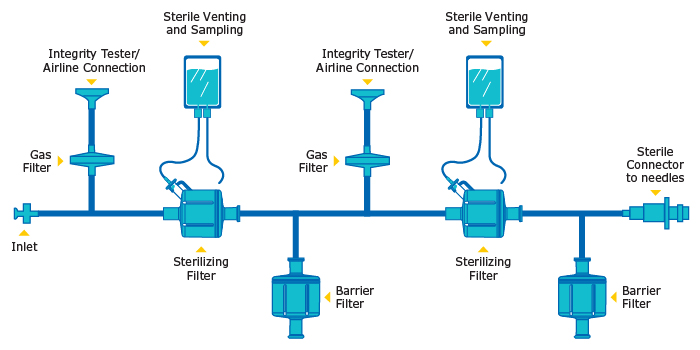
The compressed gas for filter drying should be of appropriate purity (e.g., oil free) and, once filtered, its microbiological and particle quality should be equal to or better than that of the air in the environment into which the gas is introduced.4 Oil in the air can cause contamination of the product, or hydrophobic spots on the filter, resulting in a false integrity failure. Drying can take from 15 minutes to several hours depending on the pore rating of the membrane filter, the surface area, and the set pressure10.
Prototype testing and handling considerations
Once the SUS assembly design has been finalized, it may be tested as a prototype for handling and implementation. Special consideration must be given to how these assemblies are handled and stored as they are susceptible to leaks if mishandled. For instance, cardboard is not allowed in cleanrooms, so these assemblies are removed from their outer packaging prior to transfer to the cleanroom. Assemblies should be stored horizontally on a smooth shelf, should not be over-stacked and packaging should not be folded because this will cause creases and kinks which may lead to leaks14.
The use of a support frame is recommended for these assemblies to ease handling and reduce the risk of damage. It is easier to implement a frame for a new fill line where the equipment is in the process of being installed. A frame can be designed for retrofitting to an existing fill line, however there are challenges because the space allocated for the assembly will be limited. Increasingly, assembly frames are made of plastic, and are easily fabricated; some frames show the flow path thus reducing the possibility of incorrect component installation.
Conclusion
Final filtration assemblies are single-use technologies that have significantly changed biopharmaceutical production processes by improving efficiency and offering operational flexibility.
The design of single-use assemblies can be challenging, and different options should be considered to accommodate pre-use integrity testing.
Overall, single-use final fill assemblies lead to improved manufacturing efficiency by ensuring higher batch frequencies and less work load, while significantly reducing cross contamination risks15.
Acknowledgement
Thanks to Andrew Clutterbuck, Stuart Rolfe, Stephen Cross, Trish Greenhalgh and Marie Collongues for reviewing this paper before submission.
- BCC Research. Single-Use Technologies for Biopharmaceuticals: Global Markets. 2017; https://www.bccresearch.com/market-research/biotechnology/single-use-technologies-biopharmaceuticals-report.html
- Monge M, Sinclair A. End-to-End Deployment of Single-Use Technology in Aseptic Filling of Vaccines at GSK. BioPharm International. 2010;23(2).
- Detroy A, Jenness E, Matz C, Leykin M, Acucena WR. Single-Use Technology for Syringe Filling. BioPharm International. 2014;27(3).
- FDA. Guidance for Industry Sterile Drug Products Produced by Aseptic Processing - Current Good Manufacturing Practice. 2004; http://www.fda.gov/downloads/Drugs/.../Guidances/ucm070342.pdf.
- PDA. Technical Report No. 26 (Revise 2008): Sterilising Filtration of Liquids. PDA Journal of Science and Technology. 2008;62 Supplement(S-5).
- European Commission. EU Guidelines to Good Manufacturing Practice Medicinal Products for Human and Veterinary Use. 2008; http://ec.europa.eu/health/documents/eudralex/vol-4/index_en.htm, 4.
- European Agency for the Evaluation of Medicinal Products. Note for Guidance on Manufacture of the Finished Dosage Form. 1996; http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002916.pdf
- Mok Y, Besnard L, Love T, Lesage G, Pattnaik P. Best Practices for Critical Sterile Filter Operation: A Case Study. BioProcess International. 2016;14(5).
- Felo M, Oulundsen G, Patil R. Single-Use Redundant Filtration. BioPharm International. 2012;25(4).
- Lentine KR, Gries L, Mcinnes M, Chartier J. Preparation of Redundant, Disposable Filtration Systems. BioProcess International. 2006.
- Lesage G. KVGLA 20” Integrity test by Diffusion - Qualification. 2015.
- Ito T, Hanada T, Ono K. Case Study: Pressure Rating for Bioprocess Single Use Assembly. Paper presented at: BDP Week 20152015; Huntington Beach, CA.
- Ito T, Sokolnicki A, Love T. Pressure Rating for Bioprocess Single-Use Assemblies. BioProcess International. 2016;14(3).
- Markarian J. Using Single-Use Systems in Aseptic Fill-Finish. BioPharm International. 2016;29(5):46-47.
- Laukel M, Rogge P, Dudziak G. Disposable Downstream Processing for Clinical Manufacturing. BioProcess International. 2011.