
A Quality Validation Risk Management Approach To Establishing Sampling Plans
By Allan Marinelli, Quality Validation 360 Inc.
The biopharmaceutical industry is continuously undergoing organizational changes intended to better support the healthcare industries while maintaining quality and reducing cost. This article will illustrate how manufacturers can add value to their operations by using a quality validation risk management approach for the development of sampling plans. Establishing a quality validation risk management approach for all pharmaceutical processes would increase the probability of successful outcomes for both the companies producing the products — by helping to meet regulatory requirements — while better contributing to the quality of healthcare for patients.
In this hypothetical case example, a manufacturer recently upgraded its purified water generation system with a pretreatment generation system, in order to lower maintenance costs and improve reliability/production throughout a facility. The legacy purified water generation and distribution/storage system had been validated one year prior to the upgrade. The manufacturer set out to develop a sampling plan for the updated system during the performance qualification (PQ) phase. The following approaches were considered (not an exhaustive list):
- Sample all sample points (critical and non-critical)
- Sample only the sample points determined by a single system owner (not a subject matter expert, or SME)
- Sample only the sample points determined by a cross-functional team (involving designees from various departments who are not necessarily SMEs)
- Sample only the sample points determined by an SME (or SMEs) by endorsing a quality validation risk management approach, in conjunction with the support of a cross-functional team
Option #4 was ultimately selected. The following sections will discuss how the quality validation risk management approach was applied when establishing a sampling plan during the PQ phase of the upgraded purified water (PW) generation system, including the new pretreatment generation system.
A Brief Overview of Purified Water Systems
Purified water generation systems are classified as critical utilities required to produce water that meets current U.S. Pharmacopeia Convention (USP) requirements for formulation — or as part of the medium used to produced/manufacture pharmaceutical, biopharmaceutical, combination medical devices/biologics, or vaccine intermediates and final products (final dosage forms) post-formulation.
Per the USP 39–NF 34 monograph, purified water is defined as “water obtained by a suitable process. It is prepared from water complying with the U.S. Environmental Protection Agency National Primary Drinking Water Regulations or with the drinking water regulations of the European Union, Japan, or with the World Health Organization's Guidelines for Drinking Water Quality. It contains no added substance”.1
Introduction of microbial growth and/or biofilm is one of the most critical factors affecting the output of a purified water system through generation, distribution, and storage.2 Microbial control begins with the first component in the system, (generation) since it can affect product water quality downstream and, ultimately, at points-of-use (storage/distribution).
Quality Risk Management & Sampling Activities
Although the water purification process becomes increasingly important during the middle to final purification stages, it is also important to acquire data from incoming water (source) and during various early purification stages to better assess water quality. Moreover, acquiring additional data post-validation (operation phase) can be useful in establishing a timeframe reference and, in combination with comparative analysis against baseline data from the validation phase, for assessing the reliability of the system. The additional data can also be used to determine at what stages of the purification process potential failure could occur relative to MTBF (mean time between failure), MTTR (mean time to repair), MTTF (mean time to failure), and FIT (failure in time) — and therefore prevent unnecessarily high maintenance costs.
In addition, as stipulated in the International Conference on Harmonisation (ICH) Q9 Annex II.6, Quality Risk Management as Part of Production, quality risk management is recommended to evaluate the frequency and extent of in-process control testing.3 This can be applied to PQ of the purified water generation system to reduce sampling activities while providing measures/mitigations that the system will remain in a state of proven control.
In our case example, there are a total of 21 sampling points in the pretreatment/purified water generation system. The risk assessment indicated in Table 1 was conducted and supported by the rationale in Table 2.
Note: Table 1 is not intended to be prescriptive nor exhaustive but rather a possible approach. It is recommended that the reader conform to their company’s user requirements specification prior to conducting any quality validation risk assessments.
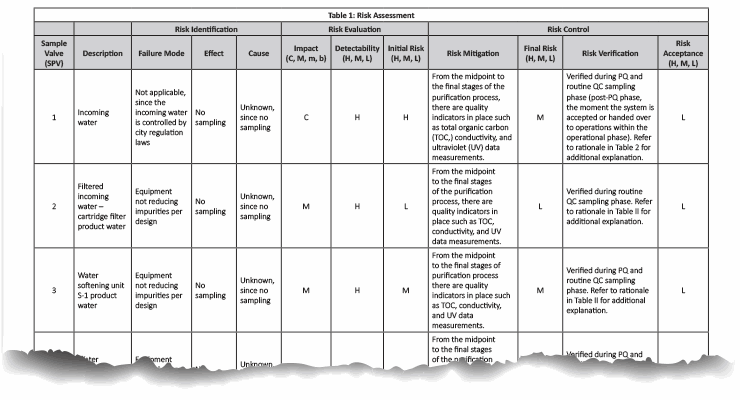
Table 1: Click on the image to view the complete table.
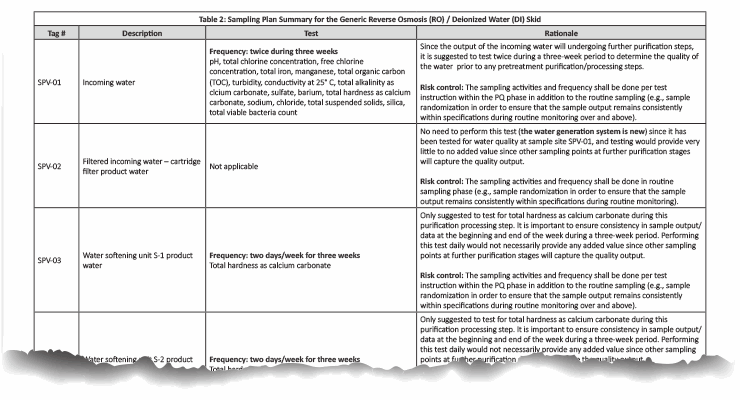
Table 2: Click on the image to view the complete table.
A potential source of harm can result from not capturing a data point that could have been used to measure the quality output, and failing to factor it in during the validation phase, only to later determine during routine monitoring that the system does not meet current USP specifications. This can result in time-consuming additional investigations and testing to account for potential anomalies that were not considered during the PQ phase.
Determining Performance Qualification (PQ) Sampling Points
In the hypothetical pretreatment/purified water generation system (skid included), there are 21 sampling points as shown in Figures 1 through 3. As part of the PQ protocol, a 21-day (minimum) intensive sampling of the purified water generation system was suggested to ensure that the water output conforms to the current USP specifications. Rather than testing all sampling points, which would have resulted in high cost (internally or externally through a third-party laboratory), a quality validation risk management approach was used to establish the sampling plan.
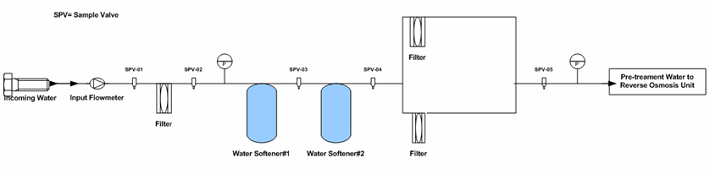
Figure 1: Example pretreatment unit sampling points (Note: The drawing is intended to illustrate where the sampling points were located, not to reflect a complete detail of the pretreatment unit.)
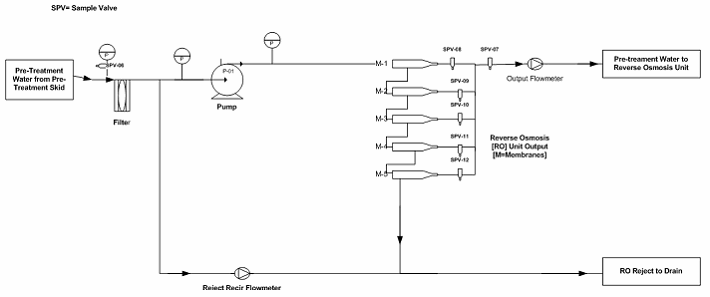
Figure 2: Example reverse osmosis (RO) unit to storage tank sampling points (Note: The drawing is intended to illustrate where the sampling points are located, not to reflect a complete detail of the RO Unit.)
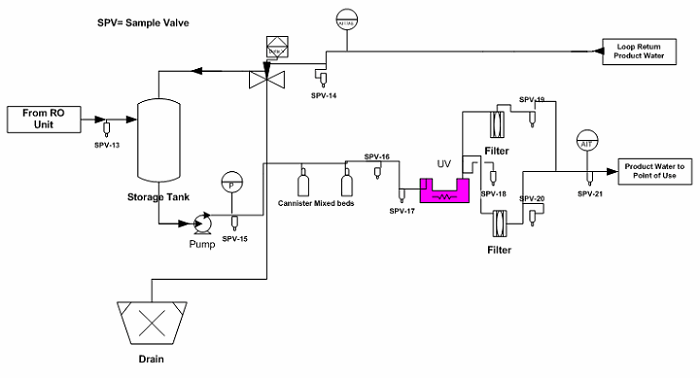
Figure 3: Example water generation system / storage tank sampling points (Note: The drawing is intended to illustrate where the sampling points are located, but not to reflect a complete detail of the water generation system / storage tank unit).
The sampling plan was constructed to entail the sample site location/frequency, the specified test, and the rationale, as shown in Table 1. The sampling plan was further adjusted and rationalized by an SME (as defined in ASTM-E2500-13 4) as shown in Table 2, supported by the risk assessment as shown in Table 1 using a qualitative approach of the failure mode effects analysis (FMEA) tool. The SME analyzed all three provided drawings (Figures 1 through 3) and concluded that only nine sampling points were needed for the system during the PQ phase for the upgraded/new purified water generation system. This inevitably resulted in a savings of about 70% of the performance qualification cost.
Conclusion
The selection of sampling points based on the SME’s (or SMEs’) guidance by effectively using the quality validation risk management approach towards the development of the sampling plan has been shown to be effective and efficient.
Collecting the following nine samples out of 21 total sampling points as part of the upgraded/new purified water generation, including the pretreatment, resulted in a saving of approximately 70 percent in validation costs:
- SPV-01: Incoming Water
- SPV-03: Water Softening Unit S-1 Product Water
- SPV-04: Water Softening Unit S-2 Product Water
- SPV-05: Activated Carbon Cartridge Filtration Product Water
- SPV-13: Reverse Osmosis Product Water
- SPV-14: USP Purified Water Distribution Loop Return
- SPV-15: USP Purified Water Storage Tank Water/Pump Discharge
- SPV-18: Loop Inline Ultraviolet Sanitization Unit Product Water
- SPV-21: USP Purified Water Distribution Loop Incoming Water
Obviously, costs would have been much higher if all sampling points were tested by a third-party laboratory, or by requiring internal laboratory QC staff to work additional hours to accommodate the analysis of validation samples over and above their already busy routine schedule.
The involvement of a subject matter expert (or experts) is crucial to the development and rationalization of the sampling plan using a quality validation risk management approach, since the results of the system have shown to be in a state of control meeting current USP specifications. In addition, this quality validation risk management approach can be easily defensible under U.S. Food and Drug Administration (FDA) regulations and those of other worldwide regulatory agencies (EMA, SFDA, KFDA, WHO, etc.).
This article is based on a chapter written by the author for the book Environmental Monitoring: A Comprehensive Handbook, Volume 7 (Chapter 9), published by PDA/DHI (2015).
References:
- USP 39-NF 34 Purified Water monograph, available at http://www.uspnf.com/
- Shnayder, Leonid, “Pharmaceutical Purified Water Storage and Distribution Systems – An Engineering Perspective”, Pharmaceutical Engineering, 21 (6), November/December 2001.
- International Conference on Harmonisation (ICH), Q9 – Quality Risk Management, Nov. 2005.
- ASTM E2500-13 – Standard Guide for Specification, Design, and Verification of Pharmaceutical and Biopharmaceutical Manufacturing Systems and Equipment, 2013.
About the Author:
 llan Marinelli, president of Quality Validation 360 Inc., has over 28 years of experience in the pharmaceutical, biopharmaceutical, medical device, vaccines, and food and beverage industries. His cGMP experience spans quality assurance (QA), compliance, quality systems, quality engineering, remediation, and validation roles controlled under FDA, EMA, and international regulations (WHO, KFDA, SFDA etc.). His experience includes, but is not limited to quality systems, CAPA coaching, remediation, QA change control coaching , QA deviation coaching, equipment validation (Writing, executing, and finalizing reports to completion), process validation, computer validation/GAMP 5, cleaning validation, quality assurance management, project management, and strategies using the ASTM-E2500-07 and ICH Q9 approaches. Allan has led clients in regulatory audit preparedness, consent decree/remediation, and FDA defense in support of facility licensure (BLA, NDA, etc.).
llan Marinelli, president of Quality Validation 360 Inc., has over 28 years of experience in the pharmaceutical, biopharmaceutical, medical device, vaccines, and food and beverage industries. His cGMP experience spans quality assurance (QA), compliance, quality systems, quality engineering, remediation, and validation roles controlled under FDA, EMA, and international regulations (WHO, KFDA, SFDA etc.). His experience includes, but is not limited to quality systems, CAPA coaching, remediation, QA change control coaching , QA deviation coaching, equipment validation (Writing, executing, and finalizing reports to completion), process validation, computer validation/GAMP 5, cleaning validation, quality assurance management, project management, and strategies using the ASTM-E2500-07 and ICH Q9 approaches. Allan has led clients in regulatory audit preparedness, consent decree/remediation, and FDA defense in support of facility licensure (BLA, NDA, etc.).
He has written chapters for numerous books published by Parenteral Drug Association (PDA) and has contributed to the International Society for Pharmaceutical Engineering’s (ISPE’s) Professional Certificate Commission (PCC), and GAMP Good Practice Guides, and Good Engineering Practices Manuals. He also published articles in the Institute of Validation Technology’s (IVT’s) Journal of Validation Technology and the American Society for Quality’s (ASQ’s) Quality Progress.
You can contact Allan at amarinelli360@gmail.com or connect with him on LinkedIn.